Expression of Human PTEN-L in a Yeast Heterologous Model Unveils Specific N-Terminal Motifs Controlling PTEN-L Subcellular Localization and Function

 , and
, and 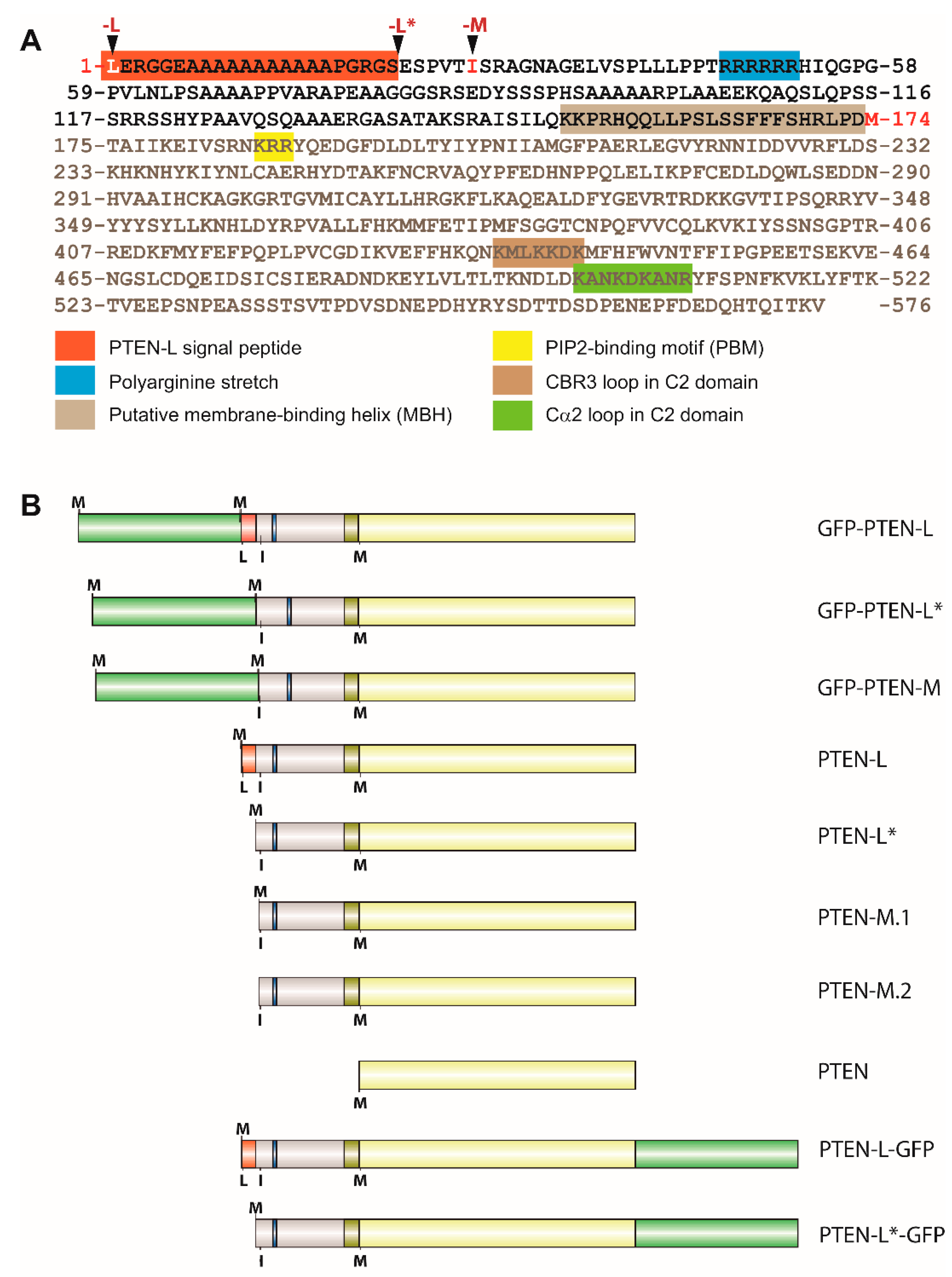{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Media and Strains
2.2. Plasmids
2.3. Fluorescence Microscopy and Image Analysis
2.4. Preparation of Cell Lysates and Immunodetection
3. Results and Discussion
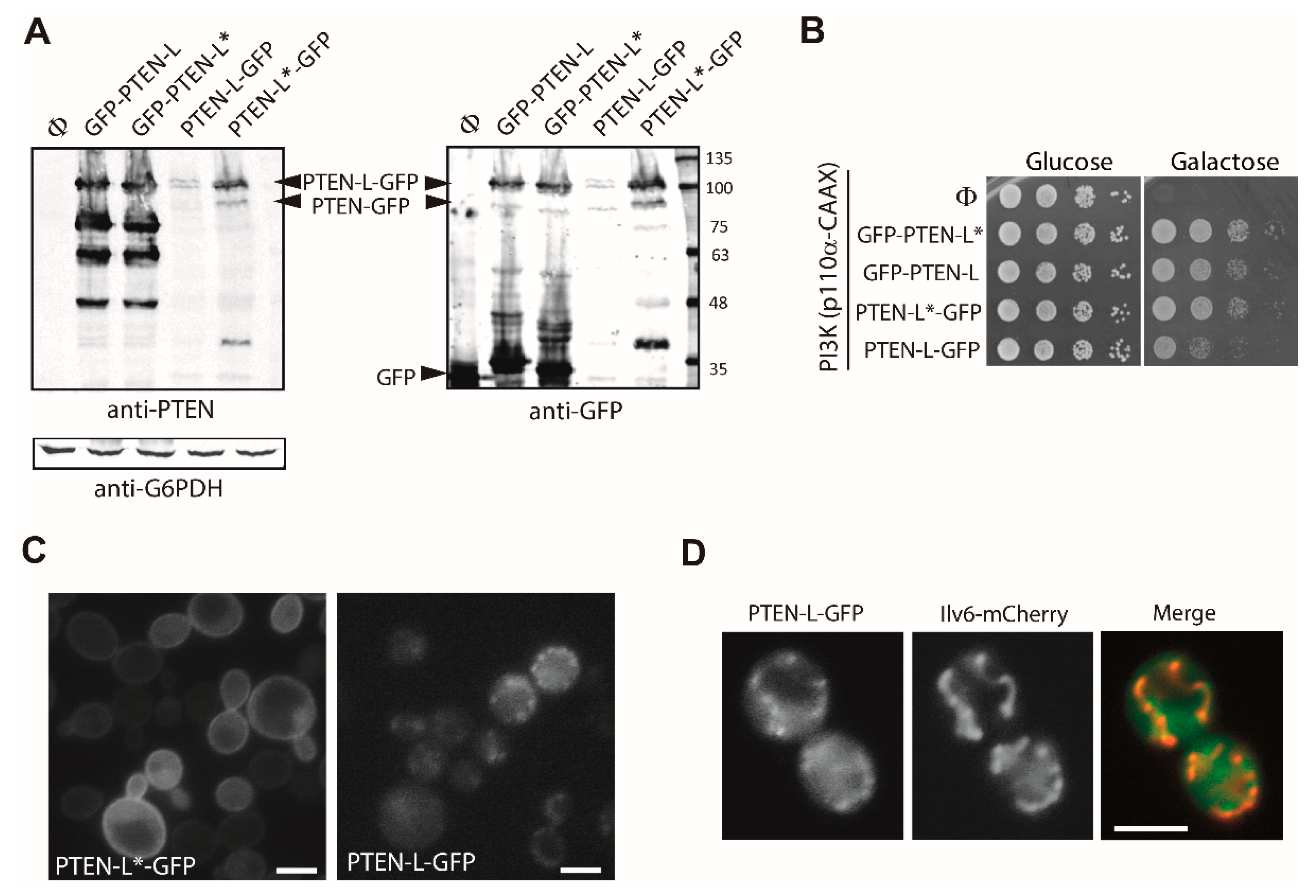
3.1. Expression of N-Terminal Extended PTEN Translational Variants in Yeast
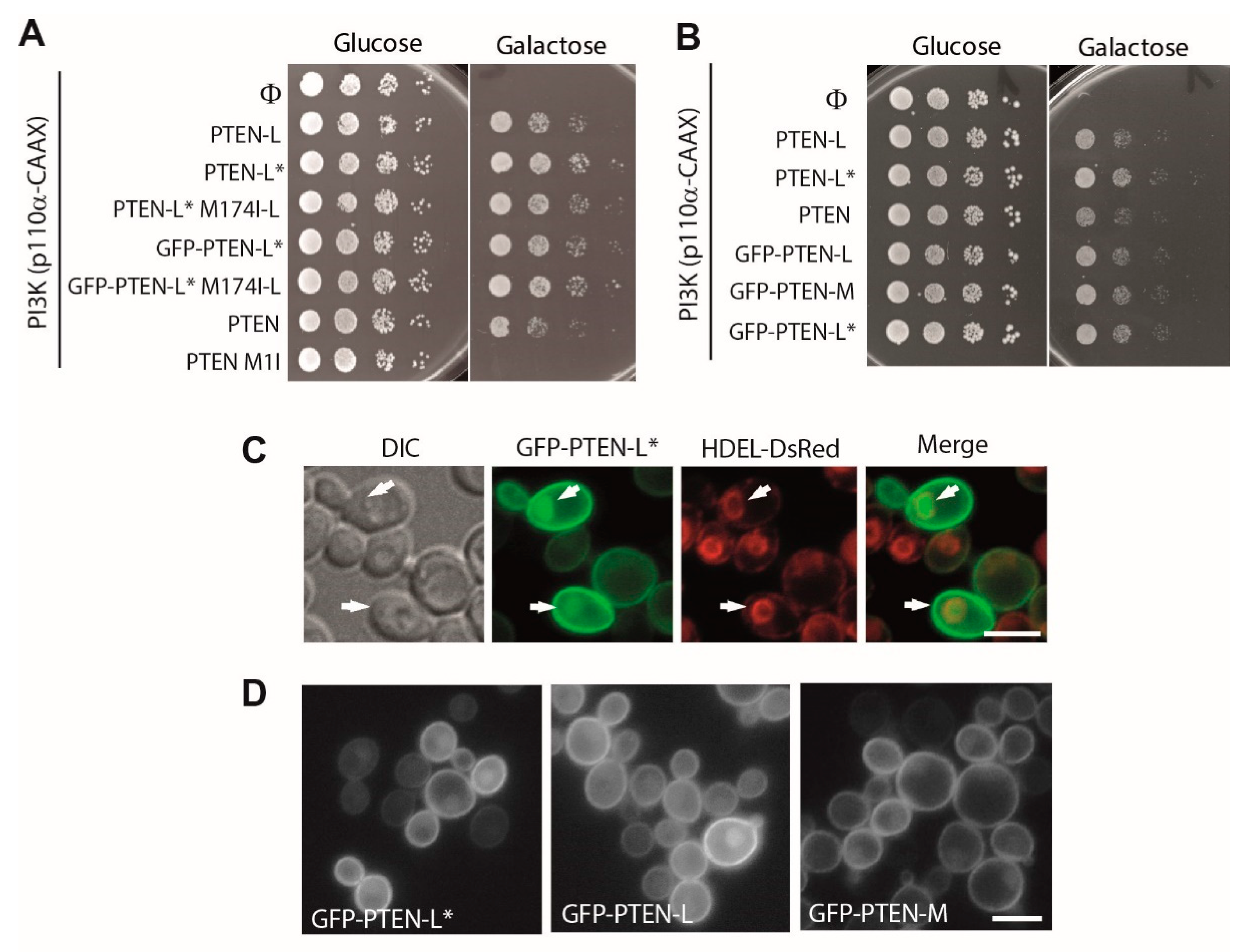
3.2. N-Terminally Extended GFP-PTEN Variants Are Functional in Yeast and Localize to the Plasma Membrane and the Nucleus
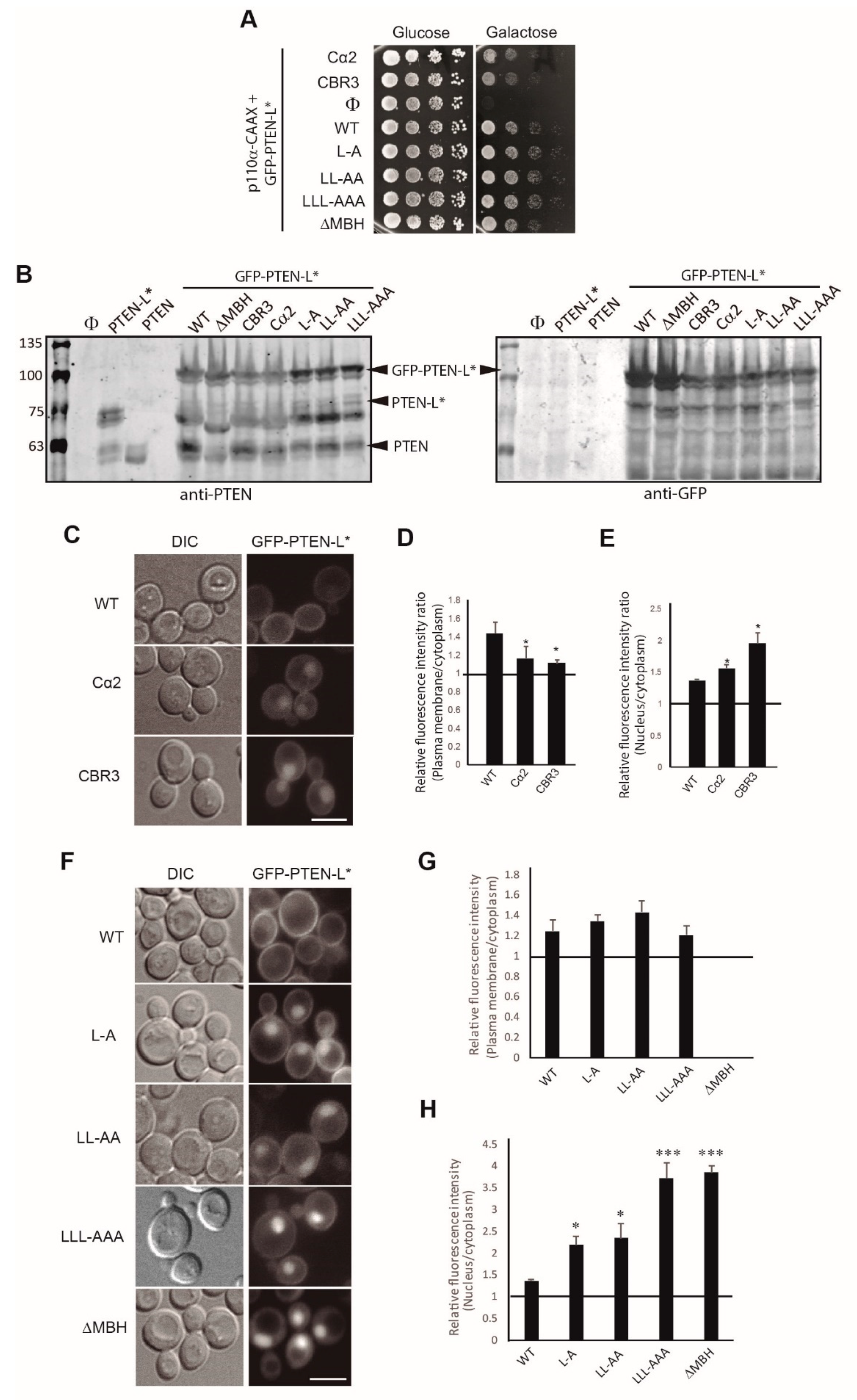
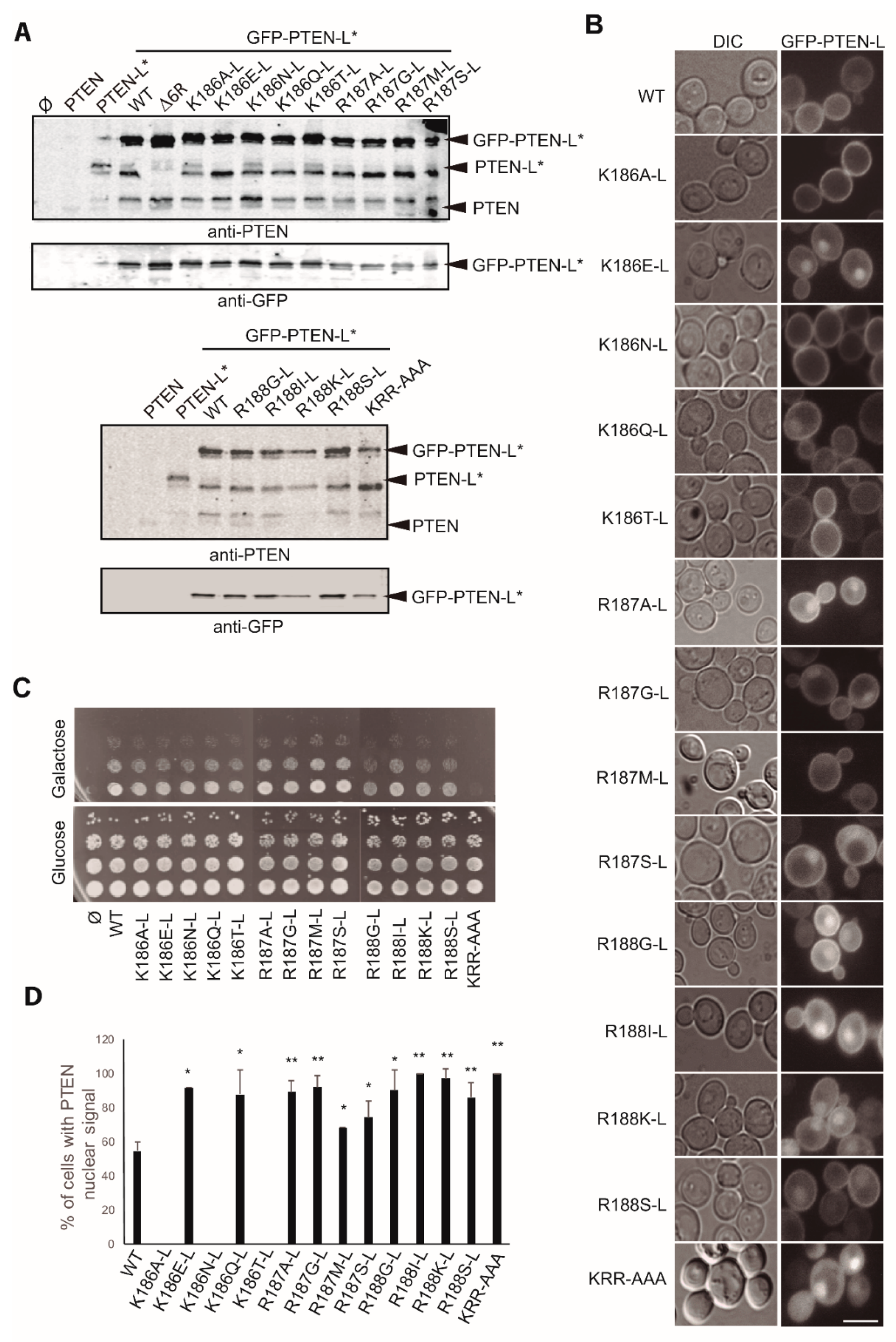
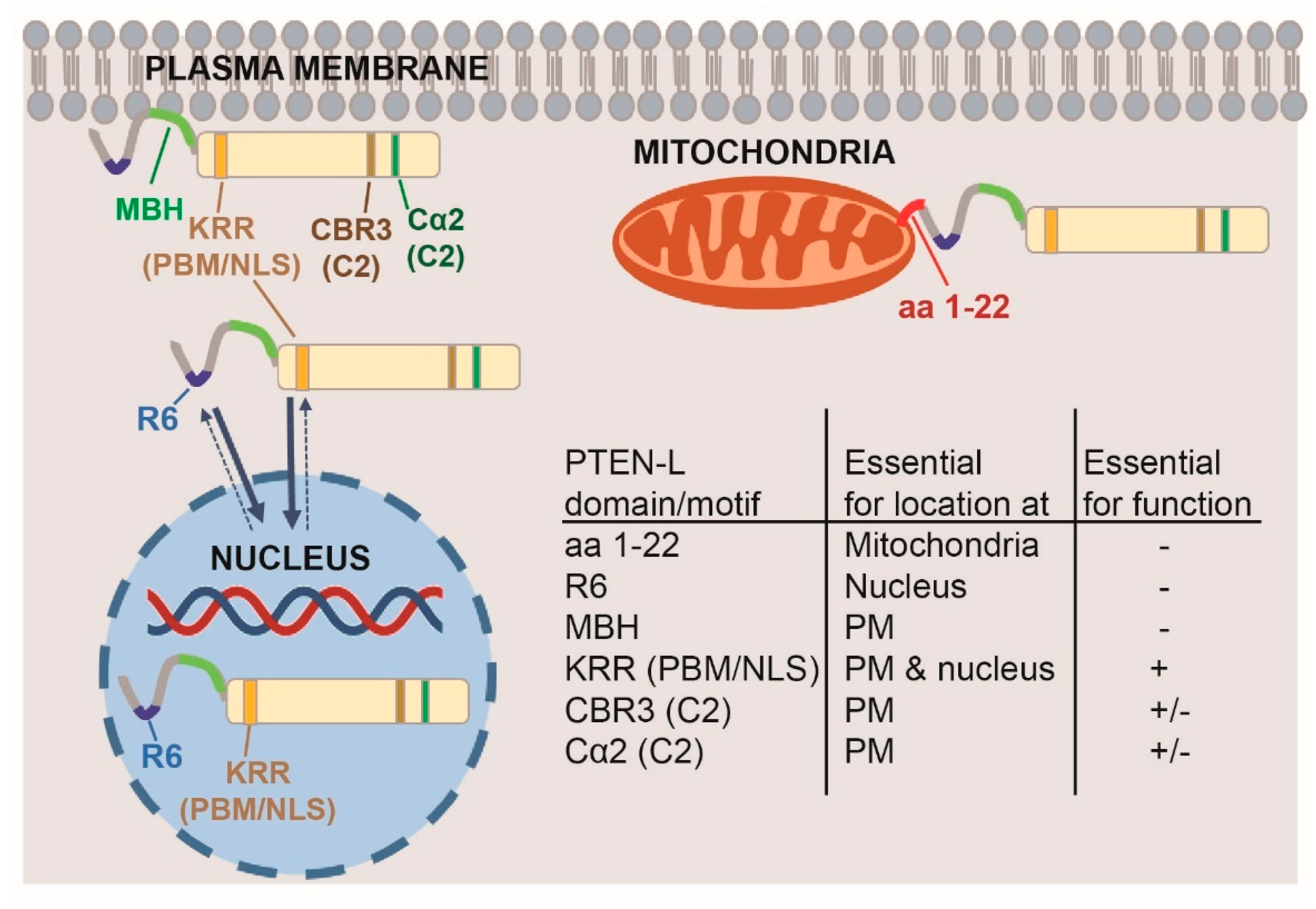
3.3. Clusters of Positively Charged and Hydrophobic Amino Acids at the C2 Domain Partially Contribute to PM-Nucleus Partition of GFP-PTEN-L in the Yeast Model
3.4. The Membrane Binding Helix at the N-Terminal Extension of PTEN-L Is Essential for Plasma Membrane Localization but not for Its Phosphatase Activity
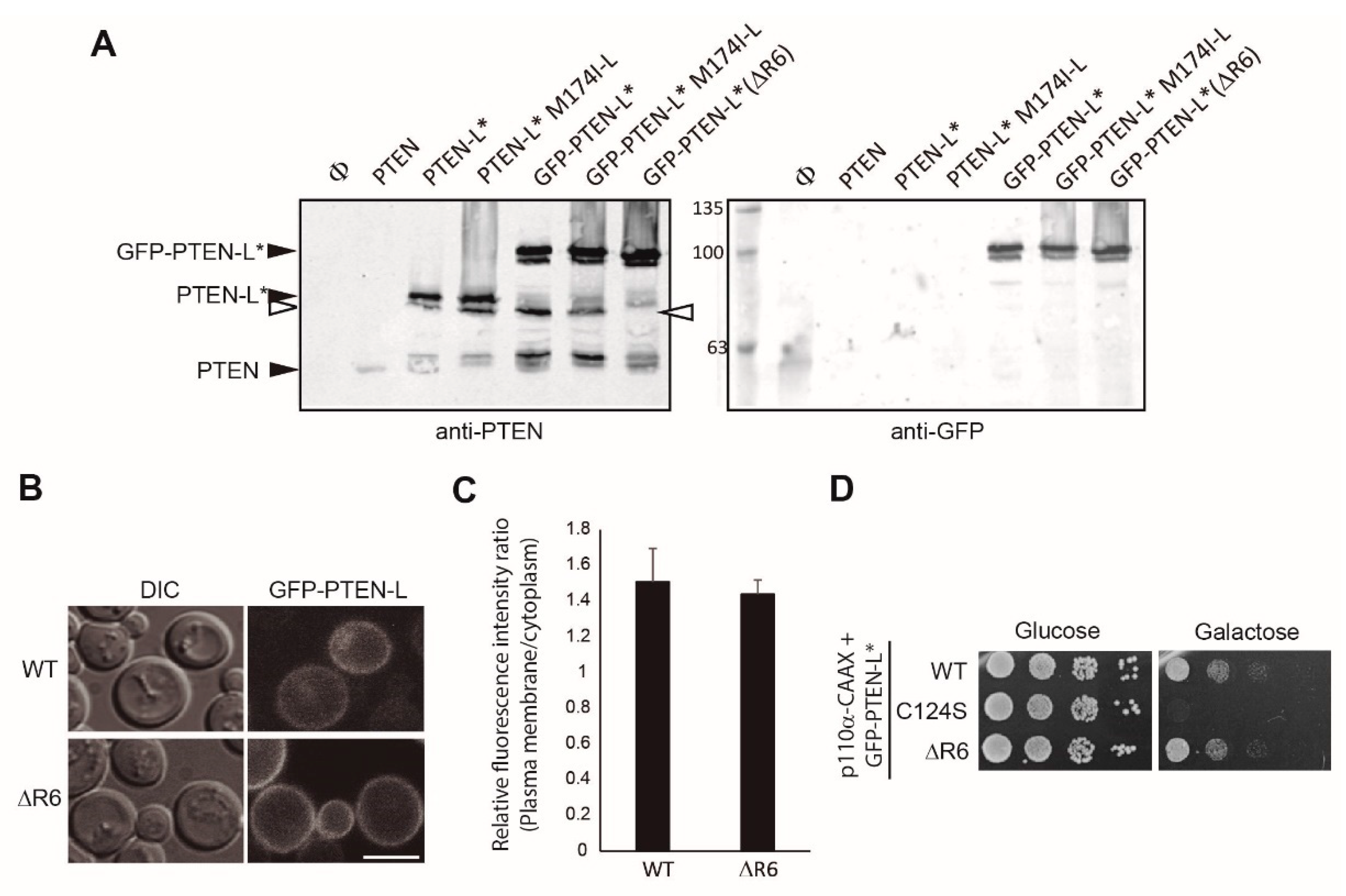
3.5. The Polyarginine Stretch in PTEN-L Is Necessary for Nuclear Localization
3.6. The Basic Cluster at the PIP2-Binding/NLS Region Regulates PTEN-L Localization at the PM and Nucleus
3.7. The First 22 Amino Acids of PTEN-L Drive the Protein to Mitochondria, Precluding Plasma Membrane Localization
Author Contributions
Funding
Conflicts of Interest
References
- Pulido, R. PTEN: A yin-yang master regulator protein in health and disease. Methods 2015, 77, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Worby, C.A.; Dixon, J.E. PTEN. Annu. Rev. Biochem. 2014, 83, 641–669. [Google Scholar] [CrossRef] [PubMed]
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Leslie, N.R.; Maccario, H.; Spinelli, L.; Davidson, L. The significance of PTEN’s protein phosphatase activity. Adv. Enzym. Regul. 2009, 49, 190–196. [Google Scholar] [CrossRef]
- Wozniak, D.J.; Kajdacsy-Balla, A.; Macias, V.; Ball-Kell, S.; Zenner, M.L.; Bie, W.; Tyner, A.L. PTEN is a protein phosphatase that targets active PTK6 and inhibits PTK6 oncogenic signaling in prostate cancer. Nat. Commun. 2017, 8, 1508. [Google Scholar] [CrossRef]
- Pulido, R. PTEN Inhibition in Human Disease Therapy. Molecules 2018, 23, 285. [Google Scholar] [CrossRef]
- Papa, A.; Pandolfi, P.P. The PTEN(-)PI3K Axis in Cancer. Biomolecules 2019, 9, 153. [Google Scholar] [CrossRef]
- Yehia, L.; Ngeow, J.; Eng, C. PTEN-opathies: From biological insights to evidence-based precision medicine. J. Clin. Invest. 2019, 129, 452–464. [Google Scholar] [CrossRef]
- Alonso, A.; Nunes-Xavier, C.E.; Bayon, Y.; Pulido, R. The Extended Family of Protein Tyrosine Phosphatases. Methods Mol. Biol. 2016, 1447, 1–23. [Google Scholar] [CrossRef]
- Myers, M.P.; Stolarov, J.P.; Eng, C.; Li, J.; Wang, S.I.; Wigler, M.H.; Parsons, R.; Tonks, N.K. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc. Natl. Acad. Sci. USA 1997, 94, 9052–9057. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.P.; Pass, I.; Batty, I.H.; Van der Kaay, J.; Stolarov, J.P.; Hemmings, B.A.; Wigler, M.H.; Downes, C.P.; Tonks, N.K. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc. Natl. Acad. Sci. USA 1998, 95, 13513–13518. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.O.; Yang, H.; Georgescu, M.M.; Di Cristofano, A.; Maehama, T.; Shi, Y.; Dixon, J.E.; Pandolfi, P.; Pavletich, N.P. Crystal structure of the PTEN tumor suppressor: Implications for its phosphoinositide phosphatase activity and membrane association. Cell 1999, 99, 323–334. [Google Scholar] [CrossRef]
- Campbell, R.B.; Liu, F.; Ross, A.H. Allosteric Activation of PTEN Phosphatase by Phosphatidylinositol 4,5-Bisphosphate. J. Biol. Chem. 2003, 278, 33617–33620. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Dixon, J.E.; Cho, W. Membrane-binding and activation mechanism of PTEN. Proc. Natl. Acad. Sci. USA 2003, 100, 7491–7496. [Google Scholar] [CrossRef]
- Denning, G.; Jean-Joseph, B.; Prince, C.; Durden, D.L.; Vogt, P.K. A short N-terminal sequence of PTEN controls cytoplasmic localization and is required for suppression of cell growth. Oncogene 2007, 26, 3930–3940. [Google Scholar] [CrossRef]
- Gil, A.; Rodriguez-Escudero, I.; Stumpf, M.; Molina, M.; Cid, V.J.; Pulido, R. A functional dissection of PTEN N-terminus: Implications in PTEN subcellular targeting and tumor suppressor activity. PLoS ONE 2015, 10, e0119287. [Google Scholar] [CrossRef]
- Gil, A.; Andrés-Pons, A.; Fernández, E.; Valiente, M.; Torres, J.; Cervera, J.; Pulido, R.; Cleveland, J. Nuclear Localization of PTEN by a Ran-dependent Mechanism Enhances Apoptosis: Involvement of an N-Terminal Nuclear Localization Domain and Multiple Nuclear Exclusion Motifs. Mol. Biol. Cell 2006, 17, 4002–4013. [Google Scholar] [CrossRef]
- Naderali, E.; Khaki, A.A.; Rad, J.S.; Ali-Hemmati, A.; Rahmati, M.; Charoudeh, H.N. Regulation and modulation of PTEN activity. Mol. Biol. Rep. 2018, 45, 2869–2881. [Google Scholar] [CrossRef]
- Liu, T.; Wang, Y.; Wang, Y.; Chan, A.M. Multifaceted Regulation of PTEN Subcellular Distributions and Biological Functions. Cancers (Basel) 2019, 11, 1247. [Google Scholar] [CrossRef]
- Yang, J.M.; Schiapparelli, P.; Nguyen, H.N.; Igarashi, A.; Zhang, Q.; Abbadi, S.; Amzel, L.M.; Sesaki, H.; Quinones-Hinojosa, A.; Iijima, M. Characterization of PTEN mutations in brain cancer reveals that pten mono-ubiquitination promotes protein stability and nuclear localization. Oncogene 2017, 36, 3673–3685. [Google Scholar] [CrossRef] [PubMed]
- Maccario, H.; Perera, N.M.; Gray, A.; Downes, C.P.; Leslie, N.R. Ubiquitination of PTEN (phosphatase and tensin homolog) inhibits phosphatase activity and is enhanced by membrane targeting and hyperosmotic stress. J. Biol. Chem. 2010, 285, 12620–12628. [Google Scholar] [CrossRef] [PubMed]
- Trotman, L.C.; Wang, X.; Alimonti, A.; Chen, Z.; Teruya-Feldstein, J.; Yang, H.; Pavletich, N.P.; Carver, B.S.; Cordon-Cardo, C.; Erdjument-Bromage, H.; et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 2007, 128, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Rahdar, M.; Inoue, T.; Meyer, T.; Zhang, J.; Vazquez, F.; Devreotes, P.N. A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc. Natl. Acad. Sci. USA 2009, 106, 480–485. [Google Scholar] [CrossRef]
- Chia, Y.C.; Catimel, B.; Lio, D.S.; Ang, C.S.; Peng, B.; Wu, H.; Zhu, H.J.; Cheng, H.C. The C-terminal tail inhibitory phosphorylation sites of PTEN regulate its intrinsic catalytic activity and the kinetics of its binding to phosphatidylinositol-4,5-bisphosphate. Arch. Biochem. Biophys. 2015, 587, 48–60. [Google Scholar] [CrossRef]
- Andres-Pons, A.; Gil, A.; Oliver, M.D.; Sotelo, N.S.; Pulido, R. Cytoplasmic p27Kip1 counteracts the pro-apoptotic function of the open conformation of PTEN by retention and destabilization of PTEN outside of the nucleus. Cell Signal 2012, 24, 577–587. [Google Scholar] [CrossRef]
- Papa, A.; Wan, L.; Bonora, M.; Salmena, L.; Song, M.S.; Hobbs, R.M.; Lunardi, A.; Webster, K.; Ng, C.; Newton, R.H.; et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 2014, 157, 595–610. [Google Scholar] [CrossRef]
- Heinrich, F.; Chakravarthy, S.; Nanda, H.; Papa, A.; Pandolfi, P.P.; Ross, A.H.; Harishchandra, R.K.; Gericke, A.; Losche, M. The PTEN Tumor Suppressor Forms Homodimers in Solution. Structure 2015, 23, 1952–1957. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, M.; Lee, J.D.; Zhang, J.; Lin, S.Y.; Fu, T.M.; Chen, H.; Ishikawa, T.; Chiang, S.Y.; Katon, J.; et al. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science 2019, 364. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Fine, B.; Steinbach, N.; Dendy, M.; Rapp, Z.; Shaw, J.; Pappas, K.; Yu, J.S.; Hodakoski, C.; Mense, S.; et al. A Secreted PTEN Phosphatase That Enters Cells to Alter Signaling and Survival. Science 2013, 341, 399–402. [Google Scholar] [CrossRef]
- Liang, H.; He, S.; Yang, J.; Jia, X.; Wang, P.; Chen, X.; Zhang, Z.; Zou, X.; McNutt, M.A.; Shen, W.H.; et al. PTENalpha, a PTEN isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism. Cell Metab. 2014, 19, 836–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulido, R.; Baker, S.J.; Barata, J.T.; Carracedo, A.; Cid, V.J.; Chin-Sang, I.D.; Dave, V.; den Hertog, J.; Devreotes, P.; Eickholt, B.J.; et al. A Unified Nomenclature and Amino Acid Numbering for Human PTEN. Sci. Signal. 2014, 7, pe15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zhang, P.; Lin, C.; Yu, Q.; Wu, J.; Wang, L.; Cui, Y.; Wang, K.; Gao, Z.; Li, H. Relevance and therapeutic possibility of PTEN-long in renal cell carcinoma. PLoS ONE 2015, 10, e114250. [Google Scholar] [CrossRef]
- Tzani, I.; Ivanov, I.P.; Andreev, D.E.; Dmitriev, R.I.; Dean, K.A.; Baranov, P.V.; Atkins, J.F.; Loughran, G. Systematic analysis of thePTEN5′ leader identifies a major AUU initiated proteoform. Open Biol. 2016, 6, 150203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masson, G.R.; Perisic, O.; Burke, J.E.; Williams, R.L. The intrinsically disordered tails of PTEN and PTEN-L have distinct roles in regulating substrate specificity and membrane activity. Biochem. J. 2015, 473, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Yang, J.; Yang, C.; Zhu, M.; Jin, Y.; McNutt, M.A.; Yin, Y. PTENalpha regulates mitophagy and maintains mitochondrial quality control. Autophagy 2018, 14, 1742–1760. [Google Scholar] [CrossRef]
- Wang, L.; Cho, Y.L.; Tang, Y.; Wang, J.; Park, J.E.; Wu, Y.; Wang, C.; Tong, Y.; Chawla, R.; Zhang, J.; et al. PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1-Parkin-mediated mitophagy. Cell Res. 2018, 28, 787–802. [Google Scholar] [CrossRef]
- Liang, H.; Chen, X.; Yin, Q.; Ruan, D.; Zhao, X.; Zhang, C.; McNutt, M.A.; Yin, Y. PTENβ is an alternatively translated isoform of PTEN that regulates rDNA transcription. Nat. Commun. 2017, 8, 14771. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Escudero, I.; Fernandez-Acero, T.; Bravo, I.; Leslie, N.R.; Pulido, R.; Molina, M.; Cid, V.J. Yeast-based methods to assess PTEN phosphoinositide phosphatase activity in vivo. Methods 2015, 77, 172–179. [Google Scholar] [CrossRef]
- Rodriguez-Escudero, I.; Roelants, F.M.; Thorner, J.; Nombela, C.; Molina, M.; Cid, V.J. Reconstitution of the mammalian PI3K/PTEN/Akt pathway in yeast. Biochem. J. 2005, 390, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Andres-Pons, A.; Rodriguez-Escudero, I.; Gil, A.; Blanco, A.; Vega, A.; Molina, M.; Pulido, R.; Cid, V.J. In vivo functional analysis of the counterbalance of hyperactive phosphatidylinositol 3-kinase p110 catalytic oncoproteins by the tumor suppressor PTEN. Cancer Res. 2007, 67, 9731–9739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, M.D.; Fernandez-Acero, T.; Luna, S.; Rodriguez-Escudero, I.; Molina, M.; Pulido, R.; Cid, V.J. Insights into the pathological mechanisms of p85alpha mutations using a yeast-based phosphatidylinositol 3-kinase model. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Escudero, I.; Oliver, M.D.; Andres-Pons, A.; Molina, M.; Cid, V.J.; Pulido, R. A comprehensive functional analysis of PTEN mutations: Implications in tumor- and autism-related syndromes. Hum. Mol. Genet. 2011, 20, 4132–4142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mighell, T.L.; Evans-Dutson, S.; O’Roak, B.J. A Saturation Mutagenesis Approach to Understanding PTEN Lipid Phosphatase Activity and Genotype-Phenotype Relationships. Am. J. Hum. Genet. 2018, 102, 943–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Acero, T.; Rodriguez-Escudero, I.; Molina, M.; Cid, V.J. The yeast cell wall integrity pathway signals from recycling endosomes upon elimination of phosphatidylinositol (4,5)-bisphosphate by mammalian phosphatidylinositol 3-kinase. Cell Signal 2015, 27, 2272–2284. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Georgescu, M.M.; Kirsch, K.H.; Kaloudis, P.; Yang, H.; Pavletich, N.P.; Hanafusa, H. Stabilization and productive positioning roles of the C2 domain of PTEN tumor suppressor. Cancer Res. 2000, 60, 7033–7038. [Google Scholar]
- Malaney, P.; Uversky, V.N.; Dave, V. PTEN proteoforms in biology and disease. Cell Mol. Life Sci. 2017, 74, 2783–2794. [Google Scholar] [CrossRef]
- Gil, A.; Andrés-Pons, A.; Pulido, R. Nuclear PTEN: A tale of many tails. Cell Death Differ. 2006, 14, 395–399. [Google Scholar] [CrossRef]
- Nguyen, H.N.; Yang, J.M.; Afkari, Y.; Park, B.H.; Sesaki, H.; Devreotes, P.N.; Iijima, M. Engineering ePTEN, an enhanced PTEN with increased tumor suppressor activities. Proc. Natl. Acad. Sci. USA 2014, 111, E2684–E2693. [Google Scholar] [CrossRef] [Green Version]
- Mingo, J.; Rodriguez-Escudero, I.; Luna, S.; Fernandez-Acero, T.; Amo, L.; Jonasson, A.R.; Zori, R.T.; Lopez, J.I.; Molina, M.; Cid, V.J.; et al. A pathogenic role for germline PTEN variants which accumulate into the nucleus. Eur. J. Hum. Genet. 2018, 26, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.B.; Raines, R.T. Catalysis by the tumor-suppressor enzymes PTEN and PTEN-L. PLoS ONE 2015, 10, e0116898. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Acero, T.; Bertalmio, E.; Luna, S.; Mingo, J.; Bravo-Plaza, I.; Rodríguez-Escudero, I.; Molina, M.; Pulido, R.; Cid, V.J. Expression of Human PTEN-L in a Yeast Heterologous Model Unveils Specific N-Terminal Motifs Controlling PTEN-L Subcellular Localization and Function. Cells 2019, 8, 1512. https://doi.org/10.3390/cells8121512
Fernández-Acero T, Bertalmio E, Luna S, Mingo J, Bravo-Plaza I, Rodríguez-Escudero I, Molina M, Pulido R, Cid VJ. Expression of Human PTEN-L in a Yeast Heterologous Model Unveils Specific N-Terminal Motifs Controlling PTEN-L Subcellular Localization and Function. Cells. 2019; 8(12):1512. https://doi.org/10.3390/cells8121512
Chicago/Turabian StyleFernández-Acero, Teresa, Eleonora Bertalmio, Sandra Luna, Janire Mingo, Ignacio Bravo-Plaza, Isabel Rodríguez-Escudero, María Molina, Rafael Pulido, and Víctor J. Cid. 2019. "Expression of Human PTEN-L in a Yeast Heterologous Model Unveils Specific N-Terminal Motifs Controlling PTEN-L Subcellular Localization and Function" Cells 8, no. 12: 1512. https://doi.org/10.3390/cells8121512