Developing Targeted Therapies That Exploit Aberrant Histone Ubiquitination in Cancer


1
Department of Biochemistry & Medical Genetics, University of Manitoba, Winnipeg, MB R3E 0J9, Canada
2
Research Institute in Oncology and Hematology, CancerCare Manitoba, Winnipeg, MB R3E 0V9, Canada
*
Author to whom correspondence should be addressed.
Cells 2019, 8(2), 165; https://doi.org/10.3390/cells8020165
Submission received: 28 January 2019
/
Revised: 12 February 2019
/
Accepted: 13 February 2019
/
Published: 16 February 2019
(This article belongs to the Special Issue Ubiquitination in Health and Disease)
Abstract
:Histone ubiquitination is a critical epigenetic mechanism regulating DNA-driven processes such as gene transcription and DNA damage repair. Importantly, the cellular machinery regulating histone ubiquitination is frequently altered in cancers. Moreover, aberrant histone ubiquitination can drive oncogenesis by altering the expression of tumor suppressors and oncogenes, misregulating cellular differentiation and promoting cancer cell proliferation. Thus, targeting aberrant histone ubiquitination may be a viable strategy to reprogram transcription in cancer cells, in order to halt cellular proliferation and induce cell death, which is the basis for the ongoing development of therapies targeting histone ubiquitination. In this review, we present the normal functions of histone H2A and H2B ubiquitination and describe the role aberrant histone ubiquitination has in oncogenesis. We also describe the key benefits and challenges associated with current histone ubiquitination targeting strategies. As these strategies are predicted to have off-target effects, we discuss additional efforts aimed at developing synthetic lethal strategies and epigenome editing tools, which may prove pivotal in achieving effective and selective therapies targeting histone ubiquitination, and ultimately improving the lives and outcomes of those living with cancer.
1. Introduction
Epigenetics is the regulation of DNA-driven processes, such as gene transcription, DNA replication, and DNA damage repair, by chromatin modifications other than changes to the DNA sequence [1]. Epigenetic mechanisms include chromatin remodeling, DNA methylation, and histone post-translational modifications, as well as the regulation of these chromatin alterations by non-coding RNAs, including microRNAs and long non-coding RNAs. Epigenetic regulation of gene expression normally governs development, cellular differentiation, and maintenance of stem-cell populations in adult tissues [2,3,4]; however, epigenetic aberrations occur in virtually all cancers and are proposed to drive oncogenesis, at least in part by repressing tumor suppressor genes and/or activating oncogenes [1,2]. Conceptually, as epigenetic alterations are inherently reversible, the therapeutic targeting and reversal of the aberrant epigenetic mechanisms in cancer cells is predicted to restore normal epigenetic regulation of gene expression, promote cell differentiation, halt cellular proliferation, and induce cell death.
For over two decades, cancer epigenetics research and therapeutic strategies focused predominantly on targeting aberrant DNA methylation and histone methylation/acetylation. To date, two DNA hypomethylating agents (azacitidine, decitabine) and four histone deacetylase inhibitors (vorinostat, romidepsin, belinostat, panobinostat) received Food and Drug Administration (FDA) approval as second- or third-line chemotherapies for certain hematological malignancies [5,6,7,8,9]. Clinical development is now focused on evaluating the relevance of these epigenetic agents for the treatment of solid tumors, as well as developing novel therapeutic strategies that target the proteins recruited to the epigenetic/chromatin modifications (known as “readers”) [9,10]. In parallel, there is an emerging interest in therapeutically targeting the machinery responsible for the addition (“writers”) or removal (“erasers”) of additional histone modifications, such as histone ubiquitination. Histone ubiquitination is of particular interest, as aberrant ubiquitination drives oncogenesis by altering the expression of key tumor suppressors and oncogenes and promoting cancer cell proliferation [11,12,13,14,15,16,17]. Therefore, there is growing impetus to develop therapies that target aberrant histone ubiquitination to induce death in cancer cells. We begin this review by first defining the normal functions ascribed to histone ubiquitination, before discussing ongoing strategies aimed at reversing aberrant histone ubiquitination, including both their benefits and limitations. Finally, we end with a discussion of three key research areas that are predicted to enable the development of histone ubiquitination-based therapies with enhanced selectivity and efficacy in cancer.
2. Histone Ubiquitination Regulates Gene Transcription and DNA Damage Repair
Ubiquitin is an 8.5-kDa polypeptide consisting of a globular domain and a flexible six-amino-acid carboxy-terminal tail terminating with a glycine residue. Ubiquitination is the formation of an isopeptide bond between this terminal glycine and the ε-amino group of a lysine residue from the target protein [18,19]. The addition of a ubiquitin moiety to a target protein is catalyzed by the successive actions of three enzymes (Figure 1). Ubiquitin is first loaded onto a ubiquitin-activating enzyme (E1), in an ATP-dependent reaction, before being transferred to a ubiquitin-conjugating enzyme (E2), and finally ligated to the target lysine residue by a ubiquitin ligase (E3) [18,20]. E3 ubiquitin ligases are primarily responsible for substrate specificity [18]. Target proteins may be monoubiquitinated or polyubiquitinated, with chains of two to ten ubiquitin molecules covalently linked via one of the seven lysine residues contained in the ubiquitin molecule or its N-terminal methionine [19]. K48-linked polyubiquitin chains generally target proteins for proteasomal degradation via the 26S proteasome, while monoubiquitin and K63-linked polyubiquitin chains typically modulate protein function, localization, or interaction with DNA and/or other proteins [19]. Protein ubiquitination levels are dynamically regulated by the opposing activity of deubiquitinating enzymes (DUBs), also known as ubiquitin-specific peptidases (USPs).
In vivo, all four core histones H2A, H2B, H3, and H4, and the linker H1 may be mono- or polyubiquitinated on multiple lysine residues [21]. These dynamic post-translational modifications control gene transcription and DNA damage repair through multiple mechanisms, including regulating histone–DNA interaction, nucleosome stability, histone eviction, chromatin compaction, histone cross-talk, or recruitment of reader proteins [22,23,24,25,26]. Unfortunately, the majority of histone ubiquitination events (i.e., specific lysine residue coupled with specific ubiquitination topology) are yet to be ascribed a biological function, particularly those found on histones H1, H3, and H4. For instance, while ring finger protein 8 (RNF8)-mediated H1 polyubiquitination plays an important role in the repair of DNA double-strand breaks (DSBs), it remains unclear which H1 lysine residues are involved in this process [27]. Similarly, while H3 and H4 ubiquitination may participate in several DNA damage repair pathways, the relevant ubiquitination sites remain to be identified [22,28]. Accordingly, this review focuses on the ubiquitination of histones H2A and H2B, whose functions and regulation are better understood (Table 1 and Table 2).
Histone H2A can be ubiquitinated on lysine residues K13, K15, K119, K125, K127, and K129 in vivo [21]. K119 is the most frequently observed ubiquitination site, and monoubiquitinated K119 (H2AK119ub1) occurs on approximately 10% of all nucleosomal H2As [29,30,31,32]. The predominant E3 ubiquitin ligase for H2AK119ub1 is the catalytic subunit of the polycomb repressive complex (PRC1), composed of really interesting new gene 1A (RING1A) and RING1B, and activated by B-lymphoma Moloney murine leukemia virus insertion region 1 homolog (BMI1) [33,34,35]. The major H2AK119ub1 DUBs are USP16 and breast cancer type 1 susceptibility protein (BRCA1)-associated protein 1 (BAP1) [36,37]. H2AK119ub1 is enriched within promoter regions of polycomb target genes and functions as a transcriptional repressor through a variety of mechanisms (Table 1) [38]. H2AK119ub1-mediated repression of polycomb target genes is necessary for the maintenance of stem-cell populations within adult tissues, including intestinal and hematopoietic stem cells, and dynamic regulation of H2AK119ub1 by USP16 and BMI1 controls normal hematopoiesis [37,39,40,41,42]. In addition, H2AK119ub1 is involved in DNA damage repair, as DSBs induce localized BMI1-mediated monoubiquitination of K119, which represses transcription of the break-flanking regions and promotes repair by homologous recombination [43,44,45,46]. In addition to H2AK119ub1, H2A ubiquitination also occurs on additional lysine residues in response to DNA damage. For example, RNF168-mediated monoubiquitination of H2AK15 is required for the recruitment of tumor protein 53 (TP53)-binding protein 1 (53BP1) to DSBs, while H2AK15 polyubiquitination (K63 linkage) by RNF168 recruits the BRCA1-A complex. Both 53BP1 and the BRCA1-A complex modulate DNA end resection [27,47,48,49,50]. In addition, BRCA1 is an important E3 ubiquitin ligase that monoubiquitinates H2AK127 and/or H2AK129 to recruit the SWI/SNF-related, matrix-associated actin-dependent regulator of chromatin, subfamily A, containing DEAD/H box 1 protein (SMARCAD1), a chromatin remodeling protein that promotes DNA end resection and homologous recombination [29,51].
As with H2A, histone H2B is ubiquitinated on multiple lysine residues in vivo, including K34 and K120. H2BK120 monoubiquitination (H2BK120ub1) is the most abundant form of H2B ubiquitination, present on approximately 1% of all nucleosomal H2Bs [32,66], and its addition is catalyzed predominantly by the E3 ubiquitin ligase complex RNF20/40 [67,68]. USP22, which is ubiquitously expressed in human tissues, is arguably the best characterized H2BK120ub1 DUB [69,70,71]. However, at least eight other DUBs can deubiquitinate H2BK120ub1 in human cells (Table 2). Although certain DUBs may be partially redundant [72], ongoing characterization of these enzymes indicate that they associate with distinct complexes, target different regions of the genome, and function in distinct pathways and cellular processes (Table 2). In addition, differences in tissue expression levels suggest that H2BK120ub1 DUBs may have tissue-specific functions [71]. To date, the complex composed of male-specific lethal 1 homolog (MSL1) and MSL2 is the only E3 ubiquitin ligase known to catalyze H2BK34 monoubiquitination (H2BK34ub1) [66,73] and a corresponding DUB is yet to be identified. H2BK120ub1 and H2BK34ub1 are co-dependent, as H2BK34ub1 promotes the recruitment of RNF20/40 and subsequent H2BK120 ubiquitination, while H2BK120ub1 promotes the recruitment of MSL1/MSL2 and subsequent H2BK34 ubiquitination [66,73]. In general, both H2BK120ub1 and H2BK34ub1 promote gene transcription (Table 2), while H2BK120ub1 is also necessary for efficient DSB repair and recruiting effectors for the two major DSB repair pathways, non-homologous end-joining and homologous recombination repair [25,74]. Remarkably, USP22-mediated H2BK120ub1 deubiquitination is also critical for efficient DSB repair through both of the major repair pathways [75,76], indicating that H2BK120ub1 levels must be dynamically regulated for efficient DSB repair.
3. Targeting Aberrant Histone Ubiquitination in Cancer
A large number of immunohistochemical analyses revealed that aberrant histone ubiquitination patterns exist in many cancer types. For example, global decreases in H2AK119ub1 levels occur in prostate cancer, while decreased H2BK120ub1 levels are frequently observed in breast, lung, and colorectal cancers relative to normal tissues [57,88,89,90]. Moreover, DNA- and RNA-sequencing data show that the genes encoding histone E3 ubiquitin ligases and DUBs are also frequently altered in cancers [91], and many of the enzymes possess tumor suppressor (e.g., BAP1 and RNF20) or oncogenic potential (e.g., BMI1 and USP22) [44,70,92,93,94], identifying possible mechanism(s) accounting for the aberrant histone ubiquitination levels observed within those cancers. While the normal and cancer-associated functions of H2AK15 and H2AK127/129 ubiquitination are only beginning to emerge (Table 3), the impact that aberrant H2AK119ub1 and H2BK120ub1 have in cancer is better defined. Indeed, clinical and molecular insight into H2AK119ub1 and H2BK120ub1 in cancer led to the development of new therapeutic strategies (discussed below) that seek to reset these modifications to effectively reprogram cellular transcription, halt cellular proliferation, and preferentially induce death in cancer cells.
3.1. Targeting Increased H2AK119ub1 Levels and BMI1 Overexpression in Hematological and Solid Malignancies
The role of the polycomb E3 ubiquitin ligase subunit RING1A/RING1B/BMI1 and H2AK119ub1 in the maintenance of stem-cell populations in adult tissues suggests it may harbor a role in the maintenance of cancer stem cells. In this regard, BMI1 is overexpressed and promotes cancer cell self-renewal in acute myeloid leukemia and several solid tumor types, such as pancreatic cancer, glioblastoma multiforme, diffuse intrinsic pontine glioma, colorectal cancer, and epithelial ovarian cancer [12,13,14,96,97,98,99,100,122]. In leukemic cells, BMI1 promotes cancer cell self-renewal via H2AK119ub1-mediated repression of key tumor suppressor genes, including the INK4A/ARF locus (Figure 2A) [12,13,14]. Interestingly, high expression of BMI1, RING1A, and/or RING1B correlates with worse overall survival in acute myeloid leukemia [91,122], suggesting that high H2AK119ub1 levels may be pathogenic. Collectively, these findings suggest that re-activation of key tumor suppressor genes following RING1A/RING1B/BMI1 inhibition may be a therapeutic strategy to inhibit cancer stem-cell proliferation and/or induce cell death (Figure 2B). In agreement with this possibility, several small-molecule inhibitors were developed, including the orally bioavailable compound PTC-596 that induces hyperphosphorylation and subsequent depletion of BMI1 [123]. In acute myeloid leukemia cell lines, PTC-596 decreases BMI1 and H2AK119ub1 levels and induces apoptosis, while it also prolongs survival in xenograft mouse models of acute myeloid leukemia [101]. In ovarian cancer models, PTC-596 administration induced apoptosis in ovarian cancer cell lines, and decreased tumor weight in orthotopic mouse models with an efficacy similar to that of cisplatin/paclitaxel, the current standard of care [123]. In 2015, a phase I clinical trial was carried out for adult patients with advanced solid tumors that reported manageable side effects [124]. Currently, two phase Ib trials are ongoing with PTC-596, either in combination with carboplatin/paclitaxel for patients with stage III–IV epithelial ovarian cancer receiving neoadjuvant chemotherapy, or in combination with radiation therapy for pediatric patients with high-grade glioma or diffuse intrinsic pontine glioma (Table 3). Thus, these pre-clinical findings combined with encouraging clinical study results highlight the potential utility of BMI1 inhibitors as clinically relevant therapeutic agents.
3.2. Exploiting Reduced H2BK120ub1 Abundance in Cancer Therapeutics
The global loss of H2BK120ub1 occurs in approximately 70% of primary breast [88] and colon cancer samples [89], and is associated with poorer patient outcomes in colon cancer [89]. Conceptually, the global depletion of H2BK120ub1 may occur as a result of reduced E3 ubiquitin ligase (e.g., RNF20/RNF40) activity and/or increased activity of the relevant DUBs (e.g., USP22). In this regard, the RNF20 promoter is hypermethylated in breast cancer [17] and reduced expression of RNF20 and/or RNF40 is observed in seminoma, basal-like breast cancer, and colorectal cancer [15,111,112]. Conversely, USP22 overexpression is associated with poor patient outcomes in multiple cancer types, including colorectal, pancreatic, breast, and epithelial ovarian cancers [89,114,115,116,117,118,119]. Reduced RNF20 expression and the concomitant reduction in H2BK120ub1 levels are known to activate the expression of key proto-oncogenes (e.g., MYC and FOS) and, therefore, may be significant drivers of oncogenesis [17]. In addition, depletion of RNF20 and H2BK120ub1 induces replication stress, adversely impacts DNA damage repair, and underlies chromosome instability, aberrant phenotypes that all contribute to cancer development and progression [111,125,126,127]. Furthermore, USP22 is a transcriptional activator which cooperates with several oncoproteins (e.g., c-MYC and the androgen receptor) to activate expression of their target genes following H2BK120ub1 deubiquitination [80,128]. Consequently, USP22 is frequently hailed as an attractive target for cancer therapeutics [89,114,115,116,117,118,119]. Unfortunately, however, a USP22-specific inhibitor remains to be identified and, thus, an attractive alternative may be to employ synthetic lethality, a strategy that selectively targets cancers with genetic defects in cancer-promoting genes. In genetic terms, synthetic lethality defines a rare and lethal combination of two independently viable mutations (Figure 3) and can be levied against cancer cells harboring specific alterations in key genes (e.g., RNF20 deletion) by downregulating the expression/function of a synthetic lethal interactor that is required for cancer cell survival. For example, RNF20-depleted cancer cells are more sensitive to DNA damage induced by the PARP1 inhibitors olaparib or BMN673 than isogenic control cells [120]. Such synthetic lethal strategies may represent an effective alternative to developing DUB inhibitors, and could be employed to target cancers exhibiting aberrant H2BK120ub1 depletion.
3.3. Increased H2BK120ub1 Abundance as an Emerging Cancer Therapeutic Target
Although the immunohistochemical studies described above show that H2BK120ub1 levels are reduced in many cancer types, there are also immunohistochemical studies showing that H2BK120ub1 levels are increased in a subset of cancers [15,88,89]. For example, whereas H2BK120ub1 levels are reduced in triple-negative breast cancers, increased H2BK120ub1 abundance occurs in luminal estrogen-receptor (ER)-positive breast cancers [15]. Perhaps even more important, high H2BK120ub1 levels are associated with worse patient survival in ER-positive breast cancer cases [15]. Furthermore, DNA- and RNA-sequencing data show that multiple cancer types, including colorectal cancer, luminal breast cancer, and ovarian cancer, frequently exhibit gene amplification or overexpression of RNF20 and/or RNF40, and/or deletion (i.e., heterozygous or homozygous deletion) and reduced expression of USP22 [15,70,91], suggesting that aberrant increases in H2BK120ub1 may also promote oncogenesis. In this regard, high H2BK120ub1 levels and RNF20/RNF40 expression promote expression of ER target genes [15,88] and proliferation of luminal breast cancer cells [15]. In mixed lineage leukemia (MLL)-rearranged leukemia, RNF20 expression also promotes cell proliferation, and RNF20-mediated H2BK120ub1 is enriched within the body of MLL-fusion target genes and promotes their expression [121]. In addition, H2BK120ub1 plays important roles in DSB repair (see Section 2 and Table 2), and reduced expression and/or function of USP22 prevents timely H2BK120ub1 deubiquitination, which impairs the DSB repair process [75,76]. Thus, reduced USP22 expression may induce chromosome instability and promote cancer development and progression. Collectively, these recent observations coupled with those detailed above (Section 3.2) suggest that H2BK120ub1 levels must be tightly regulated, as both decreases and increases likely promote oncogenesis. Accordingly, cancer patients with increased H2BK120ub1 abundance may benefit from the development of therapeutic strategies that target overexpression of H2BK120ub1 E3 ubiquitin ligases or exploit reduced DUB activity via synthetic lethality.
4. Limitations and Challenges of Therapies Targeting Histone Ubiquitination
Translation of strategies targeting histone ubiquitination into the clinic is currently hampered by four fundamental challenges. Firstly, small-molecule inhibitors against major histone E3 ubiquitin ligases and DUBs are needed, as, to date, clinically relevant inhibitors were only reported for a small subset, including RING1A/RING1B/BMI1. Technical challenges associated with the development of specific E3 ubiquitin ligase or DUB inhibitors (reviewed elsewhere [129,130]) include the lack of suitable assays reproducing physiological conditions [131,132]. In addition, DUB inhibitors directed against the catalytic thiol group are often non-specific as DUB specificity is regulated by factors independent of the catalytic site (i.e., interactions with the ubiquitin and target protein, and allosteric regulation) [133]. However, new in vitro E3 ubiquitin ligase and DUB activity assays that better model the physiological conditions and reduce false-positive rates may expedite the development of histone E3 ubiquitin ligase and DUB inhibitors [131,134,135,136]. In addition, novel cell-based assays enabling high-throughput in vivo drug screening may facilitate the identification of inhibitors with acceptable pharmacological properties [136,137]. Furthermore, better characterizing allosteric regulations of histone E3 ubiquitin ligases and DUBs may highlight new targeting opportunities. For example, the E3 ubiquitin ligase activity of RING1A/RING1B is now targeted with an inhibitor of the allosteric activator BMI1. Similarly, since the deubiquitinating activity of USP22 for H2BK120ub1 functions within the multimeric Spt–Ada–Gcn5 acetyltransferase (SAGA) complex, inhibitors could conceivably be developed that target USP22-interacting partners within the SAGA complex, such as ataxin 7 like 3 (ATXN7L3) [72]. Thus, improved screening technologies coupled with ongoing characterization of enzymes of interest are likely to yield novel inhibitors of histone E3 ubiquitin ligases and DUBs.
Secondly, although targeting histone ubiquitination based on an individual patient’s tumor biology may be considered a form of “targeted therapy”, important concerns remain regarding potential off-target effects. Similar to traditional epigenetics therapies that target DNA methylation or histone acetylation, targeting H2AK119ub1 or H2BK120ub1 will induce genome-wide transcriptional changes affecting hundreds of genes in addition to the loci of interest (Figure 2C) [13,17,61]. The impact of these changes may be largely cell-type-dependent and further modulated by the mutational landscape. This therapeutic concern is supported by a recent report indicating that RNF20 differentially regulates multiple genes in basal-like breast cancer compared to luminal breast cancer. For example, RNF20 represses the oncogene EZH2 in the basal breast cancer cell line HCC1937, while promoting its expression in the luminal breast cancer cell line MCF7 [15]. Thus, determining whether targeting H2AK119ub1 or H2BK120ub1 in a given context will promote or suppress cancer cell proliferation may be largely unpredictable and require time-consuming, individualized analysis of various cancer subtypes. This may hinder uptake of therapies targeting histone ubiquitination for treatment of additional cancer beyond those for which they are initially approved. In addition, and as with many current treatment approaches, one can expect that, within solid tumors exhibiting high levels of intratumoral genetic heterogeneity, distinct cell populations will respond differently to treatment, and proliferation of certain subpopulations could conceivably be increased by the treatment, rendering it ineffective or promoting resistance. Furthermore, if a treatment is administered systemically, transcriptional reprogramming may also occur within normal cells, and altered expression of oncogenes and tumor suppressors may result in secondary cancers. Therefore, the safety of transcription-focused therapies exploiting histone ubiquitination may be improved by ongoing efforts to design drug-delivery mechanisms that selectively deliver treatments to the targeted cancer cells (e.g., nanoparticle delivery) [138].
Thirdly, most histone ubiquitination sites are regulated by multiple E3 ubiquitin ligases and DUBs (see Table 1 and Table 2), and functional compensation may drive therapeutic resistance. Characterization of each of these enzymes is still ongoing as regards cell-type specificity, temporal dynamic, and impact on cellular processes including transcription and DNA damage repair; thus, the level of functional complementation remains unclear. If certain enzymes are fully functionally redundant, multiple E3 ubiquitin ligases or DUBs would need to be simultaneously inhibited to achieve the required therapeutic benefit. In addition, partially redundant E3 ubiquitin ligases or DUBs, with different substrate affinities or distinct temporal dynamics, may become overexpressed to compensate for target inhibition and underlie treatment resistance.
Finally, as with histone acetyltransferases, it is becoming increasingly evident that histone E3 ubiquitin ligases and DUBs can also target non-histone substrates [69,92,139]. Thus, a therapeutic strategy targeting a specific E3 ubiquitin ligase or DUB may have additional off-target effects, including misregulation of the abundance, cellular localization, and/or function of these non-histone substrates (Figure 2C). These unintended effects may be better understood in the future, as ongoing research efforts seek to identify these non-histone targets.
5. Harnessing the Full Potential of Therapies Targeting Histone Ubiquitination to Improve Outcomes of Cancer Patients
The development of therapies targeting histone ubiquitination is at an early stage of development. Broad clinical adoption of these therapies will require substantial research efforts aimed at (1) better characterizing the functions and cell-type specificity of histone E3 ubiquitin ligases and DUBs, (2) understanding the transcriptional impact of manipulating histone ubiquitination, and (3) developing selective inhibitors with appropriate pharmacological properties. In parallel, ongoing efforts to develop more selective epigenome editing tools and synthetic lethal strategies, and to characterize the role of histone ubiquitination in DNA damage repair are likely to accelerate the development of therapies targeting histone ubiquitination with enhanced specificity and efficacy.
Identifying and exploiting synthetic lethal dependencies of cancer cells with aberrant histone ubiquitination will enable the development of therapeutic strategies that bypass the potential pleiotropic off-target effects associated with direct targeting of an aberrantly expressed histone E3 ubiquitin ligase or DUB. Instead, targeting an appropriate synthetic lethal partner with a well-defined impact on a single pathway may be a more predictable and a safer strategy to exploit aberrant regulation of histone ubiquitination in cancers. Moreover, synthetic lethal strategies have the unique benefit of enabling targeting of loss-of-function alterations, which would be relevant for cancer types frequently exhibiting reduced expression of E3 ubiquitin ligases (e.g., RNF20 or RNF40) or DUBs (e.g., BAP1 or USP22).
Beyond synthetic lethal strategies, epigenome editing is an attractive alternative, as it employs fusion proteins combining a sequence-specific DNA recognition domain with the catalytic domain of a chromatin-modifying enzyme to specifically alter chromatin at selected loci (Figure 4) [140]. This technique, currently in its infancy, may enable the development of strategies modulating histone ubiquitination that target a specific locus (or loci) of interest without inducing genome-wide transcriptional changes, or altering ubiquitination levels of E3 ubiquitin ligase and DUB non-histone substrates. Employing this technique will require systematic identification of the critical driver genes aberrantly regulated by histone ubiquitination in a particular cancer type. Nevertheless, developing epigenome-editing effectors to add or remove histone ubiquitination at key loci has strong therapeutic potential that could be applied in many different cancer contexts; however, as with any novel therapeutic strategy, off-target effects would need to be adequately addressed.
Finally, although current histone ubiquitination-based strategies focus predominantly on transcriptional reprogramming of cancer cells, exploiting the role of histone ubiquitination in DNA damage repair may be an effective alternative. Importantly, clinical assessments of epigenetic therapies employing histone deacetylase inhibitors indicate that transcriptional reprogramming may not be the most effective use of these epigenetic therapies, particularly within solid tumors exhibiting genetic and epigenetic heterogeneity [2,141,142]. Importantly, emerging data show that employing histone deacetylase inhibitors to sensitize cancer cells to DNA damage-inducing treatments may be more effective and selective toward cancer cells, and better limit off-target effects and treatment resistance [2,141]. Similarly, exploiting the critical functions of histone ubiquitination in DSB repair may further sensitize cancer cells to existing DNA-damage-inducing therapies and, thus, may be very effective in a combinatorial-based therapeutic strategy. As DSB repair pathways are conserved across cell types, histone ubiquitination-based therapies that target DNA damage repair are predicted to be more broadly applicable than the inherently cell-type-dependent transcription-focused strategies. Furthermore, selective targeting of cancer cells can be achieved by targeting histone ubiquitination marks associated with DSB repair, on which cancer cells are uniquely dependent for survival (i.e., synthetic lethality). For example, upregulation of the H2AK15 E3 ubiquitin ligase RNF168 is critical for the resistance of cancer cells to endogenous or treatment-induced proteotoxic stress [95] and, therefore, RNF168 may be a promising target for a therapeutic strategy exploiting aberrant histone ubiquitination in cancers. Ongoing efforts to characterize both normal and cancer-associated functions of histone ubiquitination within the DNA damage repair pathways may highlight additional targets for the development of DNA damage-centered histone ubiquitination therapies.
6. Conclusions
Tremendous progress in our understanding of the function of histone ubiquitination in cancers is supporting the development of therapeutic strategies that aim to reprogram transcription, in order to halt cancer cell proliferation and induce cell death. However, such transcription-based strategies are currently limited by multiple challenges associated with pleiotropic off-target effects that may restrict their use to cancer types with poor prognosis and limited treatment options. These shortcomings are driving the development of new approaches, including epigenome editing tools and synthetic lethal strategies, which aim to increase the specificity, efficacy, and applicability of therapies exploiting histone ubiquitination, to ultimately benefit a broader spectrum and increased number of cancer patients.
Funding
This research was funded by the Natural Sciences and Engineering Research Council of Canada (RGPIN-05007) and the CancerCare Manitoba Foundation. Studentship funding was also received from the CancerCare Manitoba Foundation/Research Manitoba.
Acknowledgments
We thank members of the McManus laboratory and Sabine Hombach-Klonisch, Tamra Werbowetski-Ogilvie, Francis Amara and Jeff Wigle for constructive criticisms during the writing of this review. We acknowledge the strong and ongoing support of the Research Institute in Oncology and Hematology and the CancerCare Manitoba Foundation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dawson, M.A.; Kouzarides, T. Cancer Epigenetics: From Mechanism to Therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Laugesen, A.; Helin, K. Chromatin Repressive Complexes in Stem Cells, Development, and Cancer. Cell Stem Cell 2014, 14, 735–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piunti, A.; Shilatifard, A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 2016, 352, aad9780. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.-H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminskas, E.; Farrell, A.T.; Wang, Y.-C.; Sridhara, R.; Pazdur, R. FDA drug approval summary: Azacitidine (5-azacytidine, Vidaza) for injectable suspension. Oncologist 2005, 10, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Yee, A.J.; Raje, N.S. Panobinostat and Multiple Myeloma in 2018. Oncologist 2018, 23, 516–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Morschhauser, F.; Wilhelm, M.; Pinter-Brown, L.; et al. Romidepsin for the treatment of relapsed/refractory peripheral T-cell lymphoma: Pivotal study update demonstrates durable responses. J. Hematol. Oncol. 2014, 7, 11. [Google Scholar] [CrossRef]
- Bennett, R.L.; Licht, J.D. Targeting Epigenetics in Cancer. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 187–207. [Google Scholar] [CrossRef]
- Morel, D.; Almouzni, G.; Soria, J.-C.; Postel-Vinay, S. Targeting chromatin defects in selected solid tumors based on oncogene addiction, synthetic lethality and epigenetic antagonism. Ann. Oncol. 2016, 28, mdw552. [Google Scholar] [CrossRef]
- Thompson, L.L.; Guppy, B.J.; Sawchuk, L.; Davie, J.R.; Mcmanus, K.J. Regulation of chromatin structure via histone post-translational modification and the link to carcinogenesis. Cancer Metastasis Rev. 2013, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Takeuchi, M.; Negishi, M.; Oguro, H.; Ichikawa, H.; Iwama, A. Bmi1 is essential for leukemic reprogramming of myeloid progenitor cells. Leukemia 2011, 25, 1335–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdouh, M.; Facchino, S.; Chatoo, W.; Balasingam, V.; Ferreira, J.; Bernier, G. BMI1 Sustains Human Glioblastoma Multiforme Stem Cell Renewal. J. Neurosci. 2009, 29, 8884–8896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreso, A.; van Galen, P.; Pedley, N.M.; Lima-Fernandes, E.; Frelin, C.; Davis, T.; Cao, L.; Baiazitov, R.; Du, W.; Sydorenko, N.; et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat. Med. 2014, 20, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Tarcic, O.; Granit, R.Z.; Pateras, I.S.; Masury, H.; Maly, B.; Zwang, Y.; Yarden, Y.; Gorgoulis, V.G.; Pikarsky, E.; Ben-Porath, I.; et al. RNF20 and histone H2B ubiquitylation exert opposing effects in Basal-Like versus luminal breast cancer. Cell Death Differ. 2017, 24, 694–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Y.; Huo, D.; Gao, J.; Wu, H.; Ye, Z.; Liu, Z.; Zhang, K.; Shan, L.; Zhou, X.; Wang, Y.; et al. Ubiquitin ligase RNF20/40 facilitates spindle assembly and promotes breast carcinogenesis through stabilizing motor protein Eg5. Nat. Commun. 2016, 7, 12648. [Google Scholar] [CrossRef] [Green Version]
- Shema, E.; Tirosh, I.; Aylon, Y.; Huang, J.; Ye, C.; Moskovits, N.; Raver-Shapira, N.; Minsky, N.; Pirngruber, J.; Tarcic, G.; et al. The histone H2B-specific ubiquitin ligase RNF20/hBRE1 acts as a putative tumor suppressor through selective regulation of gene expression. Genes Dev. 2008, 22, 2664–2676. [Google Scholar] [CrossRef] [Green Version]
- Buetow, L.; Huang, D.T. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2016, 17, 626–642. [Google Scholar] [CrossRef] [Green Version]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Groen, E.J.N.; Gillingwater, T.H. UBA1: At the Crossroads of Ubiquitin Homeostasis and Neurodegeneration. Trends Mol. Med. 2015, 21, 622–632. [Google Scholar] [CrossRef] [Green Version]
- Tweedie-Cullen, R.Y.; Reck, J.M.; Mansuy, I.M. Comprehensive mapping of post-translational modifications on synaptic, nuclear, and histone proteins in the adult mouse brain. J. Proteome Res. 2009, 8, 4966–4982. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhai, L.; Xu, J.; Joo, H.-Y.; Jackson, S.; Erdjument-Bromage, H.; Tempst, P.; Xiong, Y.; Zhang, Y. Histone H3 and H4 Ubiquitylation by the CUL4-DDB-ROC1 Ubiquitin Ligase Facilitates Cellular Response to DNA Damage. Mol. Cell 2006, 22, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, W.A.; Li, J.; Dou, Y. Effects of histone H2B ubiquitylation on the nucleosome structure and dynamics. Nucleic Acids Res. 2018, 46, 7631–7642. [Google Scholar] [CrossRef]
- McGinty, R.K.; Kim, J.; Chatterjee, C.; Roeder, R.G.; Muir, T.W. Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature 2008, 453, 812–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyal, L.; Lerenthal, Y.; Gana-Weisz, M.; Mass, G.; So, S.; Wang, S.-Y.; Eppink, B.; Chung, Y.M.; Shalev, G.; Shema, E.; et al. Requirement of ATM-Dependent Monoubiquitylation of Histone H2B for Timely Repair of DNA Double-Strand Breaks. Mol. Cell 2011, 41, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Fierz, B.; Chatterjee, C.; McGinty, R.K.; Bar-Dagan, M.; Raleigh, D.P.; Muir, T.W. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat. Chem. Biol. 2011, 7, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorslund, T.; Ripplinger, A.; Hoffmann, S.; Wild, T.; Uckelmann, M.; Villumsen, B.; Narita, T.; Sixma, T.K.; Choudhary, C.; Bekker-Jensen, S.; et al. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 2015, 527, 389–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.M.; Bhattacharyya, A.; Simkus, C.; Vallieres, B.; Veenstra, T.D.; Zhou, M. The RAG1 V(D)J recombinase/ubiquitin ligase promotes ubiquitylation of acetylated, phosphorylated histone 3.3. Immunol. Lett. 2011, 136, 156–162. [Google Scholar] [CrossRef]
- Kalb, R.; Mallery, D.L.; Larkin, C.; Huang, J.T.J.; Hiom, K. BRCA1 is a histone-H2A-specific ubiquitin ligase. Cell Rep. 2014, 8, 999–1005. [Google Scholar] [CrossRef]
- Goldknopf, I.L.; Taylor, C.W.; Baum, R.M.; Yeoman, L.C.; Olson, M.O.; Prestayko, A.W.; Busch, H. Isolation and characterization of protein A24, a “histone like” non histone chromosomal protein. J. Biol. Chem. 1975, 250, 7182–7187. [Google Scholar] [CrossRef]
- Goldknopf, I.L.; Busch, H. Isopeptide linkage between nonhistone and histone 2A polypeptides of chromosomal conjugate-protein A24. Proc. Natl. Acad. Sci. USA 1977, 74, 864–868. [Google Scholar] [CrossRef]
- West, M.H.; Bonner, W.M. Histone 2B can be modified by the attachment of ubiquitin. Nucleic Acids Res. 1980, 8, 4671–4680. [Google Scholar] [CrossRef] [Green Version]
- Gray, F.; Cho, H.J.; Shukla, S.; He, S.; Harris, A.; Boytsov, B.; Jaremko, Ł.; Jaremko, M.; Demeler, B.; Lawlor, E.R.; Grembecka, J.; et al. BMI1 regulates PRC1 architecture and activity through homo- and hetero-oligomerization. Nat. Commun. 2016, 7, 13343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Cao, R.; Wang, M.; Myers, M.P.; Zhang, Y.; Xu, R.M. Structure of a Bmi-1-Ring1B polycomb group ubiquitin ligase complex. J. Biol. Chem. 2006, 281, 20643–20649. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef] [Green Version]
- Sahtoe, D.D.; van Dijk, W.J.; Ekkebus, R.; Ovaa, H.; Sixma, T.K. BAP1/ASXL1 recruitment and activation for H2A deubiquitination. Nat. Commun. 2016, 7, 10292. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Jones, A.E.; Yang, W.; Liu, S.; Dai, Q.; Liu, Y.; Swindle, C.S.; Zhou, D.; Zhang, Z.; Ryan, T.M.; et al. The histone H2A deubiquitinase Usp16 regulates hematopoiesis and hematopoietic stem cell function. Proc. Natl. Acad. Sci. USA 2016, 113, E51–E60. [Google Scholar] [CrossRef]
- Nakagawa, T.; Kajitani, T.; Togo, S.; Masuko, N.; Ohdan, H.; Hishikawa, Y.; Koji, T.; Matsuyama, T.; Ikura, T.; Muramatsu, M.; et al. Deubiquitylation of histone H2A activates transcriptional initiation via trans-histone cross-talk with H3K4 di- and trimethylation. Genes Dev. 2008, 22, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Chiacchiera, F.; Rossi, A.; Jammula, S.; Piunti, A.; Scelfo, A.; Ordóñez-Morán, P.; Huelsken, J.; Koseki, H.; Pasini, D. Polycomb Complex PRC1 Preserves Intestinal Stem Cell Identity by Sustaining Wnt/β-Catenin Transcriptional Activity. Cell Stem Cell 2016, 18, 91–103. [Google Scholar] [CrossRef]
- Park, I.; Qian, D.; Kiel, M.; Becker, M.W.; Pihalja, M.; Weissman, I.L.; Morrison, S.J.; Clarke, M.F. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 2003, 423, 302–305. [Google Scholar] [CrossRef]
- Tian, H.; Biehs, B.; Warming, S.; Leong, K.G.; Rangell, L.; Klein, O.D.; de Sauvage, F.J. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature 2011, 478, 255–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oguro, H.; Yuan, J.; Ichikawa, H.; Ikawa, T.; Yamazaki, S.; Kawamoto, H.; Nakauchi, H.; Iwama, A. Poised lineage specification in multipotential hematopoietic stem and progenitor cells by the polycomb protein Bmi1. Cell Stem Cell 2010, 6, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Ginjala, V.; Nacerddine, K.; Kulkarni, A.; Oza, J.; Hill, S.J.; Yao, M.; Citterio, E.; van Lohuizen, M.; Ganesan, S. BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Mol. Cell. Biol. 2011, 31, 1972–1982. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Ojo, D.; Wei, F.; Wong, N.; Gu, Y.; Tang, D.; Lin, X.; Ojo, D.; Wei, F.; Wong, N.; et al. A Novel Aspect of Tumorigenesis—BMI1 Functions in Regulating DNA Damage Response. Biomolecules 2015, 5, 3396–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chagraoui, J.; Hebert, J.; Girard, S.; Sauvageau, G. An anticlastogenic function for the Polycomb Group gene Bmi1. Proc. Natl. Acad. Sci. USA 2011, 108, 5284–5289. [Google Scholar] [CrossRef] [Green Version]
- Kakarougkas, A.; Ismail, A.; Chambers, A.L.; Riballo, E.; Herbert, A.D.; Künzel, J.; Löbrich, M.; Jeggo, P.A.; Downs, J.A. Requirement for PBAF in Transcriptional Repression and Repair at DNA Breaks in Actively Transcribed Regions of Chromatin. Mol. Cell 2014, 55, 723–732. [Google Scholar] [CrossRef]
- Ochs, F.; Somyajit, K.; Altmeyer, M.; Rask, M.-B.; Lukas, J.; Lukas, C. 53BP1 fosters fidelity of homology-directed DNA repair. Nat. Struct. Mol. Biol. 2016, 23, 714–721. [Google Scholar] [CrossRef]
- Mattiroli, F.; Vissers, J.H.A.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 Ubiquitinates K13-15 on H2A/H2AX to Drive DNA Damage Signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Scully, R.; Sobhian, B.; Xie, A.; Shestakova, E.; Livingston, D.M. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev. 2011, 25, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Sobhian, B.; Shao, G.; Lilli, D.R.; Culhane, A.C.; Moreau, L.A.; Xia, B.; Livingston, D.M.; Greenberg, R.A. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 2007, 316, 1198–1202. [Google Scholar] [CrossRef]
- Densham, R.M.; Garvin, A.J.; Stone, H.R.; Strachan, J.; Baldock, R.A.; Daza-Martin, M.; Fletcher, A.; Blair-Reid, S.; Beesley, J.; Johal, B.; et al. Human BRCA1–BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct. Mol. Biol. 2016, 23, 647–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Zhu, Q.; Wani, G.; He, J.; Wang, Q.-E.; Wani, A.A. USP3 counteracts RNF168 via deubiquitinating H2A and γH2AX at lysine 13 and 15. Cell Cycle 2014, 13, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, H.; Liu, J.; Cheruiyot, A.; Lee, J.-H.; Ordog, T.; Lou, Z.; You, Z.; Zhang, Z. USP51 deubiquitylates H2AK13,15ub and regulates DNA damage response. Genes Dev. 2016, 30, 946–959. [Google Scholar] [CrossRef] [Green Version]
- Mosbech, A.; Lukas, C.; Bekker-Jensen, S.; Mailand, N. The deubiquitylating enzyme USP44 counteracts the DNA double-strand break response mediated by the RNF8 and RNF168 ubiquitin ligases. J. Biol. Chem. 2013, 288, 16579–16587. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.-M.; Wei, L.; Lan, L.; Huen, M.S.Y. The Lys63-deubiquitylating Enzyme BRCC36 Limits DNA Break Processing and Repair. J. Biol. Chem. 2016, 291, 16197–16207. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Zhu, P.; Wang, J.; Pascual, G.; Ohgi, K.A.; Lozach, J.; Glass, C.K.; Rosenfeld, M.G. Histone H2A Monoubiquitination Represses Transcription by Inhibiting RNA Polymerase II Transcriptional Elongation. Mol. Cell 2008, 29, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Zhou, W.; Wang, J.; Puc, J.; Ohgi, K.A.; Erdjument-Bromage, H.; Tempst, P.; Glass, C.K.; Rosenfeld, M.G. A histone H2A deubiquitinase complex coordinating histone acetylation and H1 dissociation in transcriptional regulation. Mol. Cell 2007, 27, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Draker, R.; Sarcinella, E.; Cheung, P. USP10 deubiquitylates the histone variant H2A.Z and both are required for androgen receptor-mediated gene activation. Nucleic Acids Res. 2011, 39, 3529–3542. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Pfeiffer, H.; Thorne, A.; McMahon, S.B. USP22, an hSAGA subunit and potential cancer stem cell marker, reverses the polycomb-catalyzed ubiquitylation of histone H2A. Cell Cycle 2008, 7, 1522–1524. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Lang, G.; Ito, S.; Bonnet, J.; Metzger, E.; Sawatsubashi, S.; Suzuki, E.; Le Guezennec, X.; Stunnenberg, H.G.; Krasnov, A.; et al. A TFTC/STAGA Module Mediates Histone H2A and H2B Deubiquitination, Coactivates Nuclear Receptors, and Counteracts Heterochromatin Silencing. Mol. Cell 2008, 29, 92–101. [Google Scholar] [CrossRef]
- Zhang, Z.; Jones, A.E.; Wu, W.; Kim, J.; Kang, Y.; Bi, X.; Gu, Y.; Popov, I.K.; Renfrow, M.B.; Vassylyeva, M.N.; et al. Role of remodeling and spacing factor 1 in histone H2A ubiquitination-mediated gene silencing. Proc. Natl. Acad. Sci. USA 2017, 114, E7949–E7958. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.; Dienstbier, M.; Hassan, R.; Schermelleh, L.; Sharif, J.; Blackledge, N.P.; De Marco, V.; Elderkin, S.; Koseki, H.; Klose, R.; et al. Targeting polycomb to pericentric heterochromatin in embryonic stem cells reveals a role for H2AK119u1 in PRC2 recruitment. Cell Rep. 2014, 7, 1456–1470. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.; Grijzenhout, A.; Underwood, E.; Ancelin, K.; Zhang, T.; Nesterova, T.B.; Anil-Kirmizitas, B.; Bassett, A.; Kooistra, S.M.; Agger, K.; et al. Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat. Commun. 2016, 7, 13661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.T.C.; Müller, C.W.; Vermeulen, M.; Müller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 2014, 21, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Uckelmann, M.; Densham, R.M.; Baas, R.; Winterwerp, H.H.K.; Fish, A.; Sixma, T.K.; Morris, J.R. USP48 restrains resection by site-specific cleavage of the BRCA1 ubiquitin mark from H2A. Nat. Commun. 2018, 9, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Zee, B.M.; Wang, Y.; Garcia, B.A.; Dou, Y. The RING Finger Protein MSL2 in the MOF Complex Is an E3 Ubiquitin Ligase for H2B K34 and Is Involved in Crosstalk with H3 K4 and K79 Methylation. Mol. Cell 2011, 43, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Zheng, Y.; Pham, A.-D.; Mandal, S.S.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Monoubiquitination of human histone H2B: The factors involved and their roles in HOX gene regulation. Mol. Cell 2005, 20, 601–611. [Google Scholar] [CrossRef]
- Kim, J.; Guermah, M.; McGinty, R.K.; Lee, J.-S.; Tang, Z.; Milne, T.A.; Shilatifard, A.; Muir, T.W.; Roeder, R.G. RAD6-Mediated transcription-coupled H2B ubiquitylation directly stimulates H3K4 methylation in human cells. Cell 2009, 137, 459–471. [Google Scholar] [CrossRef]
- Melo-Cardenas, J.; Zhang, Y.; Zhang, D.D.; Fang, D. Ubiquitin-specific peptidase 22 functions and its involvement in disease. Oncotarget 2016, 7, 44848–44856. [Google Scholar] [CrossRef] [Green Version]
- Jeusset, L.; McManus, K. Ubiquitin Specific Peptidase 22 Regulates Histone H2B Mono-Ubiquitination and Exhibits Both Oncogenic and Tumor Suppressor Roles in Cancer. Cancers 2017, 9, 167. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Atanassov, B.S.; Mohan, R.D.; Lan, X.; Kuang, X.; Lu, Y.; Lin, K.; McIvor, E.; Li, W.; Zhang, Y.; Florens, L.; et al. ATXN7L3 and ENY2 Coordinate Activity of Multiple H2B Deubiquitinases Important for Cellular Proliferation and Tumor Growth. Mol. Cell 2016, 62, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Li, L.; Zhou, B.; Qin, Z.; Dou, Y. H2B Ubiquitylation Promotes RNA Pol II Processivity via PAF1 and pTEFb. Mol. Cell 2014, 54, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kato, A.; Kobayashi, J.; Yanagihara, H.; Sakamoto, S.; Oliveira, D.V.N.P.; Shimada, M.; Tauchi, H.; Suzuki, H.; Tashiro, S.; et al. Regulation of Homologous Recombination by RNF20-Dependent H2B Ubiquitination. Mol. Cell 2011, 41, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Haddad, D.; Li, C.; Le, M.X.; Ling, A.K.; So, C.C.; Nepal, R.M.; Gommerman, J.L.; Yu, K.; Ketela, T.; et al. The SAGA Deubiquitination Module Promotes DNA Repair and Class Switch Recombination through ATM and DNAPK-Mediated γH2AX Formation. Cell Rep. 2016, 15, 1554–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Irrazabal, T.; So, C.C.; Berru, M.; Du, L.; Lam, E.; Ling, A.K.; Gommerman, J.L.; Pan-Hammarström, Q.; Martin, A. The H2B deubiquitinase Usp22 promotes antibody class switch recombination by facilitating non-homologous end joining. Nat. Commun. 2018, 9, 1006. [Google Scholar] [CrossRef] [PubMed]
- Minsky, N.; Oren, M. The RING Domain of Mdm2 Mediates Histone Ubiquitylation and Transcriptional Repression. Mol. Cell 2004, 16, 631–639. [Google Scholar] [CrossRef]
- Li, X.S.; Trojer, P.; Matsumura, T.; Treisman, J.E.; Tanese, N. Mammalian SWI/SNF-A Subunit BAF250/ARID1 Is an E3 Ubiquitin Ligase That Targets Histone H2B. Mol. Cell. Biol. 2010, 30, 1673–1688. [Google Scholar] [CrossRef] [Green Version]
- Nicassio, F.; Corrado, N.; Vissers, J.H.A.; Areces, L.B.; Bergink, S.; Marteijn, J.A.; Geverts, B.; Houtsmuller, A.B.; Vermeulen, W.; Di Fiore, P.P.; et al. Human USP3 Is a Chromatin Modifier Required for S Phase Progression and Genome Stability. Curr. Biol. 2007, 17, 1972–1977. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Varthi, M.; Sykes, S.M.; Phillips, C.; Warzecha, C.; Zhu, W.; Wyce, A.; Thorne, A.W.; Berger, S.L.; McMahon, S.B. The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol. Cell 2008, 29, 102–111. [Google Scholar] [CrossRef]
- DeVine, T.; Sears, R.C.; Dai, M.-S. The ubiquitin-specific protease USP36 is a conserved histone H2B deubiquitinase. Biochem. Biophys. Res. Commun. 2018, 495, 2363–2368. [Google Scholar] [CrossRef] [PubMed]
- Hock, A.K.; Vigneron, A.M.; Vousden, K.H. Ubiquitin-specific peptidase 42 (USP42) functions to deubiquitylate histones and regulate transcriptional activity. J. Biol. Chem. 2014, 289, 34862–34870. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Liu, X.; Yang, J.; Li, W.; Liu, S.; Liu, X.; Yang, Z.; Ren, J.; Wang, Y.; Shan, L.; et al. Imbalance of the reciprocally inhibitory loop between the ubiquitin-specific protease USP43 and EGFR/PI3K/AKT drives breast carcinogenesis. Cell Res. 2018, 28, 934–951. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Atanassov, B.S.; Li, W.; Zhang, Y.; Florens, L.; Mohan, R.D.; Galardy, P.J.; Washburn, M.P.; Workman, J.L.; Dent, S.Y.R. USP44 Is an Integral Component of N-CoR that Contributes to Gene Repression by Deubiquitinating Histone H2B. Cell Rep. 2016, 17, 2382–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Jones, A.; Joo, H.-Y.; Zhou, D.; Cao, Y.; Chen, S.; Erdjument-Bromage, H.; Renfrow, M.; He, H.; Tempst, P.; et al. USP49 deubiquitinates histone H2B and regulates cotranscriptional pre-mRNA splicing. Genes Dev. 2013, 27, 1581–1595. [Google Scholar] [CrossRef] [Green Version]
- Shiloh, Y.; Shema, E.; Moyal, L.; Oren, M. RNF20-RNF40: A ubiquitin-driven link between gene expression and the DNA damage response. FEBS Lett. 2011, 585, 2795–2802. [Google Scholar] [CrossRef] [Green Version]
- Pavri, R.; Zhu, B.; Li, G.; Trojer, P.; Mandal, S.; Shilatifard, A.; Reinberg, D. Histone H2B Monoubiquitination Functions Cooperatively with FACT to Regulate Elongation by RNA Polymerase II. Cell 2006, 125, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Prenzel, T.; Begus-Nahrmann, Y.; Kramer, F.; Hennion, M.; Hsu, C.; Gorsler, T.; Hintermair, C.; Eick, D.; Kremmer, E.; Simons, M.; et al. Estrogen-dependent gene transcription in human breast cancer cells relies upon proteasome-dependent monoubiquitination of histone H2B. Cancer Res. 2011, 71, 5739–5753. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhu, L.; Guo, T.; Wang, Y.; Yang, J. Decreased H2B monoubiquitination and overexpression of ubiquitin-specific protease enzyme 22 in malignant colon carcinoma. Hum. Pathol. 2015, 46, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Urasaki, Y.; Heath, L.; Xu, C.W. Coupling of Glucose Deprivation with Impaired Histone H2B Monoubiquitination in Tumors. PLoS ONE 2012, 7, e36775. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Seshasayee, D.; Noubade, R.; French, D.M.; Liu, J.; Chaurushiya, M.S.; Kirkpatrick, D.S.; Pham, V.C.; Lill, J.R.; Bakalarski, C.E.; et al. Loss of the Tumor Suppressor BAP1 Causes Myeloid Transformation. Science 2012, 337, 1541–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo-Cardenas, J.; Xu, Y.; Wei, J.; Tan, C.; Kong, S.; Gao, B.; Montauti, E.; Kirsammer, G.; Licht, J.D.; Yu, J.; et al. USP22 deficiency leads to myeloid leukemia upon oncogenic Kras activation through a PU.1-dependent mechanism. Blood 2018, 132, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Chroma, K.; Mistrik, M.; Moudry, P.; Gursky, J.; Liptay, M.; Strauss, R.; Skrott, Z.; Vrtel, R.; Bartkova, J.; Kramara, J.; et al. Tumors overexpressing RNF168 show altered DNA repair and responses to genotoxic treatments, genomic instability and resistance to proteotoxic stress. Oncogene 2017, 36, 2405–2422. [Google Scholar] [CrossRef] [PubMed]
- Proctor, E.; Waghray, M.; Lee, C.J.; Heidt, D.G.; Yalamanchili, M.; Li, C.; Bednar, F.; Simeone, D.M. Bmi1 Enhances Tumorigenicity and Cancer Stem Cell Function in Pancreatic Adenocarcinoma. PLoS ONE 2013, 8, e55820. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Sui, L.; Xin, T. Correlations of BMI-1 expression and telomerase activity in ovarian cancer tissues. Exp. Oncol. 2008, 30, 70–74. [Google Scholar] [PubMed]
- Senthil Kumar, S.; Sengupta, S.; Lee, K.; Hura, N.; Fuller, C.; DeWire, M.; Stevenson, C.B.; Fouladi, M.; Drissi, R. BMI-1 is a Potential Therapeutic Target in Diffuse Intrinsic Pontine Glioma. Oncotarget 2017, 8, 62962–62975. [Google Scholar] [CrossRef]
- Rizo, A.; Olthof, S.; Han, L.; Vellenga, E.; de Haan, G.; Schuringa, J.J. Repression of BMI1 in normal and leukemic human CD34+ cells impairs self-renewal and induces apoptosis. Blood 2009, 114, 1498–1505. [Google Scholar] [CrossRef] [Green Version]
- Lessard, J.; Sauvageau, G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 2003, 423, 255–260. [Google Scholar] [CrossRef]
- Nishida, Y.; Maeda, A.; Kim, M.J.; Cao, L.; Kubota, Y.; Ishizawa, J.; AlRawi, A.; Kato, Y.; Iwama, A.; Fujisawa, M.; et al. The novel BMI-1 inhibitor PTC596 downregulates MCL-1 and induces p53-independent mitochondrial apoptosis in acute myeloid leukemia progenitor cells. Blood Cancer J. 2017, 7, e527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.O.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [PubMed]
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011, 43, 668–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peña-Llopis, S.; Vega-Rubín-de-Celis, S.; Liao, A.; Leng, N.; Pavía-Jiménez, A.; Wang, S.; Yamasaki, T.; Zhrebker, L.; Sivanand, S.; Spence, P.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 2012, 44, 751–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaFave, L.M.; Béguelin, W.; Koche, R.; Teater, M.; Spitzer, B.; Chramiec, A.; Papalexi, E.; Keller, M.D.; Hricik, T.; Konstantinoff, K.; et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat. Med. 2015, 21, 1344–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Greenberg, R.A. Deciphering the BRCA1 Tumor Suppressor Network. J. Biol. Chem. 2015, 290, 17724–17732. [Google Scholar] [CrossRef] [Green Version]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [Green Version]
- Chernikova, S.B.; Razorenova, O.V.; Higgins, J.P.; Sishc, B.J.; Nicolau, M.; Dorth, J.A.; Chernikova, D.A.; Kwok, S.; Brooks, J.D.; Bailey, S.M.; et al. Deficiency in mammalian histone H2B ubiquitin ligase Bre1 (Rnf20/Rnf40) leads to replication stress and chromosomal instability. Cancer Res. 2012, 72, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Tarcic, O.; Pateras, I.S.; Cooks, T.; Shema, E.; Kanterman, J.; Ashkenazi, H.; Boocholez, H.; Hubert, A.; Rotkopf, R.; Baniyash, M.; et al. RNF20 Links Histone H2B Ubiquitylation with Inflammation and Inflammation-Associated Cancer. Cell Rep. 2016, 14, 1462–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glinsky, G.V.; Berezovska, O.; Glinskii, A.B. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J. Clin. Investig. 2005, 115, 1503–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.L.; Yang, Y.M.; Xu, H.; Dong, X.S. Increased expression of ubiquitin-specific protease 22 can promote cancer progression and predict therapy failure in human colorectal cancer. J. Gastroenterol. Hepatol. 2010, 25, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.-D.; Cui, B.-B.; Sun, L.; Zheng, H.; Huang, Q.; Tong, J.-X.; Zhang, Q.-F. The Co-expression of USP22 and BMI-1 May Promote Cancer Progression and Predict Therapy Failure in Gastric Carcinoma. Cell Biochem. Biophys. 2011, 61, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Ning, Z.; Gao, W.; Ling, J.; Wang, A.; Luo, H.; Liang, Y.; Yan, Q.; Wang, Z. Ubiquitin-specific protease 22-induced autophagy is correlated with poor prognosis of pancreatic cancer. Oncol. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Z.; Li, Y. USP22 nuclear expression is significantly associated with progression and unfavorable clinical outcome in human esophageal squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2012, 138, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yao, L.; Zhang, X.; Ji, H.; Wang, L.; Sun, S.; Pang, D. Elevated expression of USP22 in correlation with poor prognosis in patients with invasive breast cancer. J. Cancer Res. Clin. Oncol. 2011, 137, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Shi, H.; Xie, Y.; Zhao, Z.; Li, S.; Chang, C.; Cheng, X.; Li, Y. Ubiquitin specific protease 22 promotes cell proliferation and tumor growth of epithelial ovarian cancer through synergy with transforming growth factor beta1. Oncol. Rep. 2015, 33, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Guppy, B.J.; McManus, K.J. Synthetic lethal targeting of RNF20 through PARP1 silencing and inhibition. Cell. Oncol. 2017, 40, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Kawaoka, S.; Yu, M.; Shi, J.; Ni, T.; Yang, W.; Zhu, J.; Roeder, R.G.; Vakoc, C.R. Histone H2B ubiquitin ligase RNF20 is required for MLL-rearranged leukemia. Proc. Natl. Acad. Sci. USA 2013, 110, 3901–3906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saudy, N.S.; Fawzy, I.M.; Azmy, E.; Goda, E.F.; Eneen, A.; Abdul Salam, E.M. BMI1 gene expression in myeloid leukemias and its impact on prognosis. Blood Cells Mol. Dis. 2014, 53, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Xiong, X.; Crim, A.; Dwivedi, S.K.D.; Mustafi, S.B.; Mukherjee, P.; Cao, L.; Sydorenko, N.; Baiazitov, R.; Moon, Y.-C.; et al. Evaluating the Mechanism and Therapeutic Potential of PTC-028, a Novel Inhibitor of BMI-1 Function in Ovarian Cancer. Mol. Cancer Ther. 2018, 17, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Bedard, P.L.; Shapiro, G.; Bauer, T.M.; Prawira, A.; Laskin, O.; Weetall, M.; Baird, J.; Branstrom, A.; Mara, E.O.; et al. Phase 1 results of PTC596, a novel small molecule targeting cancer stem cells (CSCs) by reducing levels of BMI1 protein. ASCO Annu. Meet. Abstr. 2017, 35, 2574. [Google Scholar] [CrossRef]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.X.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, P.A.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.M.; Howell, M.; et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.; Jeusset, L.; Lepage, C.; McManus, K. Evolving Therapeutic Strategies to Exploit Chromosome Instability in Cancer. Cancers 2017, 9, 151. [Google Scholar] [CrossRef]
- Schrecengost, R.S.; Dean, J.L.; Goodwin, J.F.; Schiewer, M.J.; Urban, M.W.; Stanek, T.J.; Sussman, R.T.; Hicks, J.L.; Birbe, R.C.; Draganova-Tacheva, R.A.; et al. USP22 regulates oncogenic signaling pathways to drive lethal cancer progression. Cancer Res. 2014, 74, 272–286. [Google Scholar] [CrossRef]
- Harrigan, J.A.; Jacq, X.; Martin, N.M.; Jackson, S.P. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov. 2017, 17, 57–78. [Google Scholar] [CrossRef]
- Landré, V.; Rotblat, B.; Melino, S.; Bernassola, F.; Melino, G. Screening for E3-ubiquitin ligase inhibitors: Challenges and opportunities. Oncotarget 2014, 5, 7988–8013. [Google Scholar] [CrossRef]
- Ritorto, M.S.; Ewan, R.; Perez-Oliva, A.B.; Knebel, A.; Buhrlage, S.J.; Wightman, M.; Kelly, S.M.; Wood, N.T.; Virdee, S.; Gray, N.S.; et al. Screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nat. Commun. 2014, 5, 4763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrigley, J.D.; Eckersley, K.; Hardern, I.M.; Millard, L.; Walters, M.; Peters, S.W.; Mott, R.; Nowak, T.; Ward, R.A.; Simpson, P.B.; et al. Enzymatic Characterisation of USP7 Deubiquitinating activity and Inhibition. Cell Biochem. Biophys. 2011, 60, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Sowa, M.E.; Bennett, E.J.; Gygi, S.P.; Harper, J.W. Defining the human deubiquitinating enzyme interaction landscape. Cell 2009, 138, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Orcutt, S.J.; Wu, J.; Eddins, M.J.; Leach, C.A.; Strickler, J.E. Bioluminescence assay platform for selective and sensitive detection of Ub/Ubl proteases. Biochim. Biophys. Acta 2012, 1823, 2079–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jong, A.; Merkx, R.; Berlin, I.; Rodenko, B.; Wijdeven, R.H.M.; El Atmioui, D.; Yalçin, Z.; Robson, C.N.; Neefjes, J.J.; Ovaa, H. Ubiquitin-based probes prepared by total synthesis to profile the activity of deubiquitinating enzymes. Chembiochem 2012, 13, 2251–2258. [Google Scholar] [CrossRef]
- Harrigan, J.; Jacq, X. Monitoring Target Engagement of Deubiquitylating Enzymes Using Activity Probes: Past, Present, and Future; Humana Press: New York, NY, USA, 2016; pp. 395–410. [Google Scholar]
- Herman, A.G.; Hayano, M.; Poyurovsky, M.V.; Shimada, K.; Skouta, R.; Prives, C.; Stockwell, B.R. Discovery of Mdm2-MdmX E3 ligase inhibitors using a cell-based ubiquitination assay. Cancer Discov. 2011, 1, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.H.; Park, K. Targeted drug delivery to tumors: Myths, reality and possibility. J. Control. Release 2011, 153, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Kungulovski, G.; Jeltsch, A. Epigenome Editing: State of the Art, Concepts, and Perspectives. Trends Genet. 2016, 32, 101–113. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef]
- Ramachandran, S.; Ient, J.; Göttgens, E.-L.; Krieg, A.J.; Hammond, E.M. Epigenetic Therapy for Solid Tumors: Highlighting the Impact of Tumor Hypoxia. Genes 2015, 6, 935–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Schematic presenting the enzymatic machinery catalyzing the addition and removal of histone ubiquitination. Ubiquitination requires a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin ligase (E3). As depicted, histones are typically ubiquitinated on lysine residues contained within histone tails that extend away from the nucleosome. Ubiquitin (Ub) is removed from histones (i.e., target proteins) by a deubiquitinating enzyme (DUB).
Figure 1.
Schematic presenting the enzymatic machinery catalyzing the addition and removal of histone ubiquitination. Ubiquitination requires a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin ligase (E3). As depicted, histones are typically ubiquitinated on lysine residues contained within histone tails that extend away from the nucleosome. Ubiquitin (Ub) is removed from histones (i.e., target proteins) by a deubiquitinating enzyme (DUB).

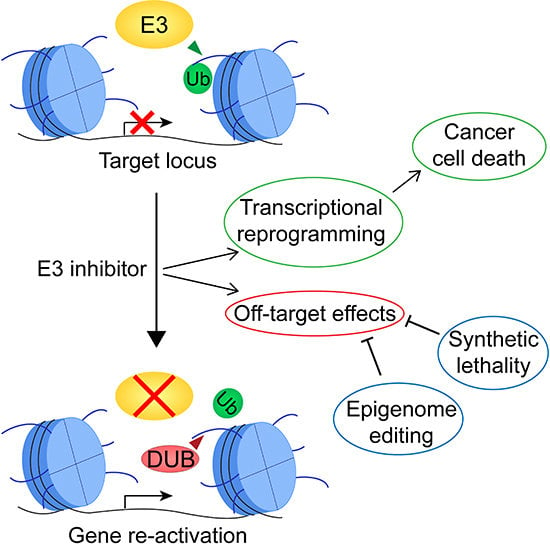
Figure 2.
Schematic presenting putative impacts associated with targeting the histone ubiquitination machinery. (A) In cancer, overexpression of a histone E3 ubiquitin ligase (e.g., really interesting new gene 1A/1B (RING1A/RING1B)) or its allosteric activator (AA; e.g., B-lymphoma Mo-MLV insertion region 1 homolog (BMI1)) can repress expression of tumor suppressor genes. (B )Following therapeutic inhibition of an E3 ubiquitin ligase or its allosteric activator, ongoing DUB activity will remove the ubiquitin mark at the locus of interest resulting in gene derepression (i.e., gene re-activation). (C) Inhibition of the E3 ubiquitin ligase impacts additional processes; it may re-activate (i), or repress expression of additional off-target genes (ii), while other genes of interest may not be re-activated if a functionally redundant E3 ubiquitin ligase compensates for the loss of the inhibited E3 ubiquitin ligase (iii). In addition, inhibition of the E3 ubiquitin ligase may deactivate additional complexes it associates with (hexagons), resulting in misregulation of ubiquitination on non-histone targets (iv). This may impact their localization and function (such as activation of transcription factors) and induce further off-target effects.
Figure 2.
Schematic presenting putative impacts associated with targeting the histone ubiquitination machinery. (A) In cancer, overexpression of a histone E3 ubiquitin ligase (e.g., really interesting new gene 1A/1B (RING1A/RING1B)) or its allosteric activator (AA; e.g., B-lymphoma Mo-MLV insertion region 1 homolog (BMI1)) can repress expression of tumor suppressor genes. (B )Following therapeutic inhibition of an E3 ubiquitin ligase or its allosteric activator, ongoing DUB activity will remove the ubiquitin mark at the locus of interest resulting in gene derepression (i.e., gene re-activation). (C) Inhibition of the E3 ubiquitin ligase impacts additional processes; it may re-activate (i), or repress expression of additional off-target genes (ii), while other genes of interest may not be re-activated if a functionally redundant E3 ubiquitin ligase compensates for the loss of the inhibited E3 ubiquitin ligase (iii). In addition, inhibition of the E3 ubiquitin ligase may deactivate additional complexes it associates with (hexagons), resulting in misregulation of ubiquitination on non-histone targets (iv). This may impact their localization and function (such as activation of transcription factors) and induce further off-target effects.

Figure 3.
The synthetic lethal paradigm in cancer targeting. Synthetic lethality describes a rare and lethal genetic interaction occurring between two unlinked genes. Mutually exclusive mutations or alterations occurring in either GENE 1 or GENE 2 are viable, while simultaneous co-occurrence is lethal. In a therapeutic context, cancer cells with genetic defects in a histone E3 ubiquitin ligase or DUB gene (e.g., GENE1) can be selectively targeted and killed by downregulating or inhibiting the expression and/or function of a synthetic lethal interactor (GENE 2; drug target).
Figure 3.
The synthetic lethal paradigm in cancer targeting. Synthetic lethality describes a rare and lethal genetic interaction occurring between two unlinked genes. Mutually exclusive mutations or alterations occurring in either GENE 1 or GENE 2 are viable, while simultaneous co-occurrence is lethal. In a therapeutic context, cancer cells with genetic defects in a histone E3 ubiquitin ligase or DUB gene (e.g., GENE1) can be selectively targeted and killed by downregulating or inhibiting the expression and/or function of a synthetic lethal interactor (GENE 2; drug target).

Figure 4.
Schematic depicting an epigenome editing tool to target histone ubiquitination. Epigenome editing employs a fusion protein consisting of a DNA sequence-specific recognition domain (gray box) and a functional chromatin-modifying domain, such as a histone E3 ubiquitin ligase, in order to modulate a chromatin post-translational modification at a specific locus.
Figure 4.
Schematic depicting an epigenome editing tool to target histone ubiquitination. Epigenome editing employs a fusion protein consisting of a DNA sequence-specific recognition domain (gray box) and a functional chromatin-modifying domain, such as a histone E3 ubiquitin ligase, in order to modulate a chromatin post-translational modification at a specific locus.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Functions and regulation of histone H2A ubiquitination marks.
| Writers 1 | Erasers | Readers and Function |
|---|---|---|
| H2AK15ub1 | ||
| RNF168 (recruited by RNF8-mediated H1 polyubiquitination) [27,48] | -USP3 [52] -USP51 [53] May also be directly or indirectly regulated by: -USP44 [54] | DNA damage repair: In the vicinity of a DNA double-strand break (DSB), H2AK15ub1 mediates recruitment of 53BP1 (in conjunction with H4K20 di-methylation). 53BP1 acts as a protein scaffold recruiting DNA repair proteins, which modulate DNA end resection and promote error-free repair [47]. |
| H2AK15 polyubiquitination (K63-linked ubiquitin chains) | ||
| Initiated by RNF168, elongated by RNF8 [48] | BRCC36 [55] | DNA damage repair: Following DSB, H2AK15 polyubiquitination recruits the BRCA1-A complex (including E3 ubiquitin ligase BRCA1/BARD1), which inhibits DNA end resection [49,50]. |
| H2AK119ub1 | ||
| -RING1A/RING1B (catalytic subunit of PRC1 complexes) activated by BMI1 [33,34,35] -DZIP3 [56] | -BAP1, activated by ASXL1, ASLX2 or ASLX3 [36] -MYSM1 [57] -USP3 (DNA damage repair) [52] -USP10 (H2A.Z variant) [58] -USP16 (regulation of hematopoiesis) [37] -USP21 [38] -USP22 [59,60] | Transcriptional repression: -Prevents recruitment of the FACT (facilitates chromatin transcription) complex, which blocks transcription elongation [56] -Prevents methylation of gene-activating marks H3K4me2-3 [38] -Recruits chromatin remodeling protein RSF1 [61] -Recruits PRC2 polycomb repressive complexes, which catalyze the repressive mark H3K27me3 [62,63,64] -Deubiquitination by MYSM1 promotes androgen receptor-dependent gene activation [57] DNA damage response: -Represses transcription of DSB-flanking regions [46] -Promotes rapid DSB repair [46] |
| H2AK127ub1 and H2AK129ub1 | ||
| BRCA1/BARD1 [29] | USP48 [65] | DNA damage response: Recruits SMARCAD1, which promotes DNA end resection and homologous recombination [51] |
1 Only E3 ubiquitin ligases, primarily responsible for substrate specificity, are listed.
Table 2.
Functions and regulation of histone H2B ubiquitination marks.
| Writers 1 | Erasers | Readers and Function |
|---|---|---|
| H2BK34ub1 | ||
| MSL1/MSL2 [66,73] | Unknown | Transcription activation: -Promotes methylation of transcription-activating marks H3K4me3 and H3K79me2 [66] -Promotes conversion of nucleosomes into hexasomes via H2A/H2B dimer eviction [23] |
| H2BK120ub1 | ||
| -RNF20/RNF40 [67,68] -MDM2 (H2BK120 activity may be restricted to p53 target genes) [77] -BAF250/Elongin-C/Cullin-2/RBX1 [78] | -USP3 (DNA damage repair) [79] -USP22 (transcription regulation; DSB repair) [60,80] -USP27X (transcription regulation) [72] -USP36 (transcriptional repression of target genes) [81] -USP42 (transcriptional activation of target genes) [82] -USP43 (transcriptional repression of NuRD target genes) [83] -USP44 (transcriptional repression of N-CoR target genes) [84] -USP49 (control of mRNA splicing) [85] -USP51 (transcription regulation) [72] | Transcription activation: -Promotes accessible chromatin conformation [26,86] -Promotes transcription elongation by RNA polymerase II in cooperation with FACT [87] -Promotes methylation of transcription-activating marks H3K4me2-3 (by SET1 complex) [68] and H3K79me1-2 (by DOT1L) [24] Transcriptional repression: -MDM2-mediated H2BK120ub1 may promote transcription repression [77] DNA damage repair: -Recruits effectors of the two major DSB repair pathways, non-homologous end-joining and homologous recombination [25,74] -Removal required for class-switch recombination repair, non-homologous end-joining and homologous recombination [75,76] |
1 Only E3 ubiquitin ligases, primarily responsible for substrate specificity, are listed.
Table 3.
Functions of histone H2A and H2B ubiquitination marks in cancer.
| Alteration of Writers or Erasers in Cancer | Clinical Development |
|---|---|
| H2AK15ub1 and H2AK15 poly-ub | |
| Proteotoxic stress (endogenous or treatment-induced) depletes ubiquitin pools available for DNA damage signaling. Overexpression of RNF168 and subsequent alteration of DSB repair processes promotes resistance to proteotoxic stress in cancer cells [95]. | Not applicable |
| H2AK119ub1 | |
| BMI1, the activator of the RING1A/RING1B E3 ubiquitin ligase, is overexpressed and promotes cancer cell self-renewal in multiple cancer types, including pancreatic cancer, glioblastoma multiforme, pediatric diffuse intrinsic pontine glioma, colorectal cancer, epithelial ovarian cancer, and acute myeloid leukemia [12,13,14,96,97,98,99,100]. BMI1 promotes self-renewal of leukemic cells in part via H2AK119ub1-mediated repression of the INK4A/ARF locus [12]. In glioblastoma and colorectal cancer, the effect of BMI1 overexpression on cancer cell self-renewal is independent of the INK4A/ARF locus and involves repression of distinct genes [13,14]. | BMI1 inhibitor PTC-596: -Preclinical studies for acute myeloid leukemia [101] -Phase I for advanced solid tumors (NCT02404480 complete) -Phase Ib in combination with carboplatin/paclitaxel for ovarian cancer patients (NCT03206645) -Phase Ib in combination with radiation for pediatric diffuse intrinsic pontine glioma or high-grade glioma (NCT03605550) |
| Reduced expression of BAP1 (DUB) occurs frequently in metastatic uveal melanoma, pleural mesothelioma, and clear-cell renal cell carcinoma [92,102,103,104]. BAP1 germline mutations are associated with a familial syndrome of predisposition to mesothelioma and uveal and cutaneous melanoma [92,105]. Relevance of aberrant H2AK119ub1 in BAP1-deficient cancers is not established. | BAP1-deficient cells may be targeted with an EZH2 inhibitor via a synthetic lethal strategy [106]. EZH2 inhibitor tazemetostat is in phase II clinical trial for patients with BAP1-deficient malignant mesothelioma (NCT02860286). |
| H2AK127ub1 and H2AK129ub1 | |
| BRCA1, which is frequently deficient in breast and ovarian cancer, is a well-established tumor suppressor protein maintaining genome integrity via its multiple roles in DNA damage repair [107]. Recent identification of BRCA1 as the E3 ubiquitin ligase for H2AK127/129 indicates that misregulation of H2AK127/129ub1 may contribute to genome instability in cancer [29]. | BRCA1 deficiency sensitizes cancer cells to synthetic lethal targeting with PARP1 inhibitors [108,109]. Advanced BRCA1-deficient ovarian cancers are treated with PARP1 inhibitors such as olaparib [110]. |
| H2BK120ub1 | |
| Global loss of H2BK120ub1 is observed in ~70% of breast and colorectal cancers [88,89]. RNF20 promoter is hypermethylated in breast cancer and RNF20/RNF40 expression is reduced in seminoma, basal-like breast cancer, and colorectal cancer [15,17,111,112]. USP22 overexpression is part of the “death from cancer” signature [113] and observed in multiple cancer types, including breast cancer and colorectal cancer [89,114,115,116,117,118,119] | Preclinical study indicates that RNF20-depleted cells can be selectively targeted with PARP1 inhibitor via a synthetic lethal strategy [120] |
| RNF20 promotes breast luminal tumor growth and RNF20-mediated H2BK120ub1 promotes expression of estrogen receptor-α target genes in luminal breast cancer cells [15]. RNF20 expression is required for proliferation of MLL rearrangement-driven leukemia [121] | Not applicable |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jeusset, L.M.-P.; McManus, K.J. Developing Targeted Therapies That Exploit Aberrant Histone Ubiquitination in Cancer. Cells 2019, 8, 165. https://doi.org/10.3390/cells8020165
AMA Style
Jeusset LM-P, McManus KJ. Developing Targeted Therapies That Exploit Aberrant Histone Ubiquitination in Cancer. Cells. 2019; 8(2):165. https://doi.org/10.3390/cells8020165
Chicago/Turabian StyleJeusset, Lucile M-P, and Kirk J McManus. 2019. "Developing Targeted Therapies That Exploit Aberrant Histone Ubiquitination in Cancer" Cells 8, no. 2: 165. https://doi.org/10.3390/cells8020165
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.