Combined Targeted Omic and Functional Assays Identify Phospholipases A2 that Regulate Docking/Priming in Calcium-Triggered Exocytosis


1
Molecular Physiology and Molecular Medicine Research Group, School of Medicine, Western Sydney University, Campbelltown Campus, NSW 2560, Australia
2
Department of Health Sciences, Faculty of Applied Health Sciences and Department of Biological Sciences, Faculty of Mathematics & Science, Brock University, St. Catharines, ON L2S 3A1, Canada
*
Author to whom correspondence should be addressed.
Cells 2019, 8(4), 303; https://doi.org/10.3390/cells8040303
Submission received: 24 February 2019
/
Revised: 24 March 2019
/
Accepted: 28 March 2019
/
Published: 2 April 2019
(This article belongs to the Special Issue 25 Years of Proteomics in Cell Biology)
Abstract
:The fundamental molecular mechanism underlying the membrane merger steps of regulated exocytosis is highly conserved across cell types. Although involvement of Phospholipase A2 (PLA2) in regulated exocytosis has long been suggested, its function or that of its metabolites—a lyso-phospholipid and a free fatty acid—remain somewhat speculative. Here, using a combined bioinformatics and top-down discovery proteomics approach, coupled with lipidomic analyses, PLA2 were found to be associated with release-ready cortical secretory vesicles (CV) that possess the minimal molecular machinery for docking, Ca2+ sensing and membrane fusion. Tightly coupling the molecular analyses with well-established quantitative fusion assays, we show for the first time that inhibition of a CV surface calcium independent intracellular PLA2 and a luminal secretory PLA2 significantly reduce docking/priming in the late steps of regulated exocytosis, indicating key regulatory roles in the critical step(s) preceding membrane merger.

1. Introduction
Fusion is a ubiquitous fundamental cellular process enabling merger of biological membranes; this includes the defining release step of exocytosis following merger of secretory vesicles with the plasma membrane (PM). Studies over the last three-four decades have suggested a role of Phospholipase A2 (PLA2) in regulated exocytosis, generally arising from correlations of arachidonic acid (ARA) levels and vesicle content release [1,2,3,4,5,6,7,8]. In a cell free system, fusion of secretory vesicles with target membranes increased in the presence of PLA2 suggesting that the enzyme or its metabolites affected membrane merger or the steps immediately preceding it [5,6,9]. Furthermore, addition of exogenous PLA2 metabolites inhibits hemi-fusion formation or transition from hemi-fusion to pore opening depending upon their site of incorporation [10,11,12]. That PLA2 species have been identified in secretory vesicles isolated from neutrophils [13], chromaffin cells [14], insulin secreting cells [15,16] and mast cells [17,18] highlights that these are conserved protein components of secretory vesicles. Early studies also identified endogenous PLA2 activity in purified synaptic vesicles (SV) [5,6,19,20]. While some proteomic analyses have failed to identify SV associated PLA2 [21,22], suggesting very low abundance and/or activity, at least one study identified iPLA2γ in synaptosomes [23]. Indeed, when synaptosomes were fractionated into free and docked SV pools prior to proteomic analysis, a patatin-like phospholipase domain containing 8 (Gene ID 157819923; also known as iPLA2γ) was found to be more abundant in the docked SV pool [24].
Based on subcellular localization and Ca2+ requirement, PLA2 isozymes are broadly classified as (i) Ca2+-dependent secretory or sPLA2, (ii) Ca2+-independent cytoplasmic or membrane bound iPLA2 and (iii) Ca2+-dependent cytoplasmic or cPLA2. Nevertheless, all PLA2 cleave membrane phospholipids at the sn-2 position, producing a lyso-phospholipid (often lyso-phosphatidylcholine; LPC) which is preferentially retained in the membrane and a diffusible free fatty acid (FFA; often arachidonic acid (ARA; 20:4n-6)) that can incorporate back into the membrane via Lands cycle intermediates or is further metabolized to yield eicosanoids. sPLA2 are generally 12–19 kDa proteins containing 5–7 conserved disulphide bonds and are characterized by an active site histidine in a DXCCXXHD consensus sequence and a conserved Ca2+-binding loop with a XCGXGG consensus sequence [25,26]. Once secreted, their Ca2+ requirement for catalysis is in the low millimolar (mM) range with no strict fatty acid selectivity, although mostly target phosphatidyl-ethanolamine, -glycerol, -inositol and -serine (PE, PG, PI and PS) [26,27]. Although these enzymes are generally thought to be active only when released into the extracellular space, the possibility of basal sPLA2 activity in the vesicle lumen has been suggested [17,28,29]. In transfected CHO-K1 and HEK293 cells that lack regulated secretory vesicles, expressed sPLA2 was localized to the golgi and related intracellular vesicles and released ARA, indicating basal activity [28]. Of note is that adequate concentrations of free calcium ([Ca2+]free) may be (transiently) present within regulated secretory vesicles to support luminal sPLA2 activity under basal conditions [30,31,32,33]. In contrast, iPLA2 are intracellular enzymes that do not require Ca2+ beyond basal cytosolic levels for activity, contain serine in the GXSXG active site and possess a nucleotide-binding (GXGXXG) consensus sequence. The mammalian forms include iPLA2β, iPLA2γ, iPLA2δ, iPLA2ε, iPLA2ζ and iPLA2η with molecular weights ranging from 27 to 146 kDa. A well-established function of iPLA2β is membrane phospholipid remodelling via Lands cycle to maintain membrane integrity [29,34,35]. cPLA2 are cytosolic proteins of molecular weights ranging from 61-114 kDa and require micromolar [Ca2+]free for association with membrane phospholipids via their N-terminal C2 domain [25,26]. These enzymes also carry serine at GXSXG, GXSGS or GXSXV lipase consensus sequences [36]. There are six mammalian cPLA2 isoforms, cPLA2-α, β, γ, δ, ε, ζ of which all except cPLA2γ have a N-terminal C2 domain [36,37].
At the genomic level, sea urchin and human remain closely related (both are deuterostomes) relative to invertebrates [38,39,40]. SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins are highly conserved across species, having been identified on exocytotic vesicles from yeast to man, including the release-ready cortical vesicles (CV) isolated from unfertilized sea urchin eggs [40,41,42,43,44,45,46]. Furthermore, as CV retain the minimal molecular machinery for docking, Ca2+ sensing and membrane fusion, they are a well-established model to study the late steps of Ca2+-triggered exocytosis [42,43,45,47,48,49,50,51,52,53,54]. CV are easily isolated with high purity and yield, either endogenously docked on the PM (i.e., cell surface complexes; CSC) or free-floating [42,49,51,55]. On triggering with Ca2+, isolated CV fuse with each other in a manner indistinguishable from CV-PM fusion [42,45,49,55]. Using these fusion-ready native vesicles, we first identified catalytically active endogenous PLA2 isozymes using a combined bioinformatics and top-down discovery proteomics approach coupled with lipidomic analyses. We then asked whether these isozymes have a role in the late steps of Ca2+-triggered exocytosis. The enzymatic activities were localized to the CV lumen and vesicle surface and tightly coupled functional and molecular analyses were carried out to assess these PLA2 activities and their associated effects on fusion parameters. A modulatory role in priming and/or docking is indicated.
2. Materials and Methods
2.1. Materials
2.1.1. Assessment of PLA2 Activities and Fusion Assays
Native sea urchins (Heliocedaris tuberculata) were maintained at 7–8 °C in the local aquatic facility. CSC and CV were isolated as previously described [38,41,42,45,51,52,55]. Bromoenol lactone (BEL) and 3-(3-acetamide-1-benzyl-2-ethylindolyl-5-oxy) propane sulfonic acid (LY311727) were purchased from Sigma Aldrich (St. Louis, MO USA); 1,1,1,2,2-Pentafluoro-7-phenyl-3-heptanone (FKGK-11) was from Cayman Chemicals (Ann Arbor, MI, USA).
CSC membrane labelling was carried out using 1-oleoyl-2-{12-[(7-nitro-2-1,3-benzoxadiazol-4-yl) amino] dodecanoyl} -sn-glycero-3-phospho-choline/-ethanolamine (NBD-PC/PE). Quantification of the generated NBD-FFA and NBD-PA was done using 1-oleoyl-2-{12-[(7-nitro-2-1,3-benzoxadiazol-4-yl) amino Arachidonic acid 20:4 (NBD-ARA) and 1-oleoyl-2-{12-[(7-nitro-2-1,3-benzoxadiazol-4-yl) amino] dodecanoyl}-sn-glycero-3-phosphate (NBD-PA) as reference standards. ARA (20:4) was purchased from Matreya LLC (State college, PA, USA) while all other lipids were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Quantitative lipid analyses were carried out using High Performance Thin Layer Chromatography (HPTLC) [49,50,51,56]. Silica G-60 plates were purchased from Merck Millipore Ltd. (Darmstadt, Germany). All HPTLC solvents were minimally of analytical grade. High grade Trypsin was from Promega Corp. (Madison, WI, USA). The selective PLA2 substrate, PED6 (N-((6-(2,4-Dinitrophenyl) amino) hexanoyl)-2-(4,4-Difluoro-5,7-Dimethyl-4-Bora-3a,4a-Diaza-s-Indacene-3-Pentanoyl)-1-Hexadecanoyl-sn-Glycero-3-Phosphoethanolamine, Triethylammonium Salt) was from Invitrogen (Carlsbad, CA, USA).
2.1.2. Protein Fractionation, Gel Electrophoresis and Western Blotting
Amicon Ultra-4 centrifugal filters (3kDa cut-off) came from Merck Millipore. Constituents of the Protease inhibitor cocktail [41,57] were purchased from Sigma-Aldrich (St. Louis) and AMRESCO Inc. (Dallas, TX, USA). All electrophoresis grade chemicals used in two-dimensional gel electrophoresis (2DE) were purchased from AMRESCO Inc. 3-10NL IPG strips, tributyl phosphine and Polyvinylidene difluoride (PVDF) membrane (0.2 µM pore size) were from Bio-Rad (Hercules, CA, USA). Anti-human rabbit polyclonal iPLA2 antibody (PA5-27945) and anti-human mouse monoclonal sPLA2 antibody (ab-24498) were from Invitrogen and Abcam (Cambridge, UK), respectively. Blocking agents―non-fat dry milk powder and BSA―were from Devondale (Saputo Dairy, Australia) and Sigma Aldrich, respectively. HRP linked anti-mouse IgG (NA93IV) and anti-rabbit IgG (NA934VS) antibodies came from GE Healthcare (Buckinghamshire, UK). Lumunata Cresendo Western HRP substrate was purchased from Merck Millipore. The blots and gels were imaged using the Image Quant™ LAS 4000 Biomolecular Imager (GE Healthcare, Chicago, Il, USA).
2.2. Methods
2.2.1. Molecular Analysis to Detect CSC Associated PLA2 Activities
PLA2 activities in intact unlabelled CSC suspensions (0.5 mL, OD 0.56 ± 0.02, n = 3–14) were assessed by measuring changes in FFA following treatments with the indicated concentrations of BEL, LY311727 and FKGK-11; exploratory experiments (n = 1–2) with 40 µM BEL, 10 µM FKGK-11, 100 nM LY311727 and higher concentrations were first carried out. Only those concentrations that significantly reduced endogenous FFA relative to control were selected for further study. CSC were isolated according to established protocols [49,50,51,52] and suspended in baseline intracellular medium (BIM; 210 mM potassium glutamate, 500 mM glycine, 10 mM NaCl, 10 mM PIPES, 50 μM CaCl2, 1 mM MgCl2, 1 mM EGTA pH 6.7) supplemented with 2.5 mM ATP, 2 mM DTT and 1X protease inhibitors [41,42,43,45,51,52]. Stock solutions of inhibitors were prepared in dimethyl sulfoxide and were delivered to CSC suspensions at a final solvent concentration of <1% [48,51,52]. At the end of the incubation, CV suspensions were immediately placed on ice, diluted with 5–8 mL of ice-cold BIM and aliquoted for rapid isolation of total lipids and for immediate assay of CV-PM fusion [49,50,51].
In separate experiments, CSC were allowed to incorporate NBD-PC or -PE for 15 min prior to the 20 min inhibitor treatments. Briefly, CSC suspensions (7 mL, OD 0.56 ± 0.005, n = 3) were labelled with 5600 picomoles NBD-PC or -PE for 15 min followed by treatment with the indicated doses of inhibitors. At the end of the treatments, all aliquots were washed three times in ice-cold BIM (pH 6.7) to remove unlabelled NBD substrate and inhibitors and immediately placed on ice prior to rapid isolation of total CSC lipids and membrane proteins (see Section 2.2.4).
2.2.2. Quantitative Lipid Analyses
Total CSC or CV lipids were extracted according to Bligh and Dyer with established modifications [50,51,52,56,58]; total CV membrane proteins were also isolated from parallel aliquots by lysing CV with ice-cold PIPES (see Section 2.2.4). The resulting organic phases (and also aqueous phases in scaled-up experiments) were recovered, dried under nitrogen and suspended in CHCl3:CH3OH (2:1; v/v) for loading onto pre-conditioned silica gel 60 HPTLC plates. Sample loading was with the CAMAG automatic TLC sampler. Pre-conditioning of the plates was as previously described [50,51,52,56]. Using the CAMAG AMD 2 multi-development unit, neutral- and phospho-lipids were resolved using optimized protocols [48,49,51,52,56]. Resolved lipids were visualized on-plate by charring homogenously wetted plates with 10% cupric sulphate in 8% aqueous phosphoric acid at 145 °C for 10 min [56]; imaging was at 460 nm/605 nm (Ex/Em) using the LAS 4000 Biomolecular Imager and was analysed using MultiGauge v3.0 (Fuji Photo Film Co., Ltd. Tokyo, Japan). Quantification of endogenous CV or CSC lipids, NBD-FFA and -PA, was via calibration curves of the appropriate standards, resolved by HPTLC, in parallel with the experimental samples. To resolve neutral and phospholipids, well-established protocols were used [56]; NBD-FFA and -PA generated from NBD-PC labelled CSC were resolved together after confirming that the standard neutral lipid protocol was satisfactory for resolving both these species. To measure fluorescent signal from NBD-labelled lipids, uncharred plates were imaged at 460 nm/575 nm (Ex/Em) using the same imager and analysis software.
2.2.3. Sequence Alignments
To identify regions of homology, a sequence alignment of the putative urchin sPLA2 (W4XE93) and iPLA2 (XP_011669048.1) relative to characterized human and mouse isoforms was carried out using MEGA version 7.0.21 (free software downloaded from https://www.megasoftware.net/home) [59,60] and the ClustalW algorithm with default settings. All protein sequences, except that of the predicted urchin iPLA2, were downloaded from Uniprot (February 2019). Of 22 putative urchin sequences, one with sPLA2 characteristics (i.e., ~14kDa, likely to form intra-molecular disulphide bonds and prediction of Ca2+ as a cofactor [17,25]) was selected using the information available in Uniprot. Similarly, a putative urchin 80–85kDa iPLA2 was selected from the 44 entries on the basis of characteristic molecular weight and presence of ankyrin repeats—a feature common to iPLA2 forms across species [25,35]. As the predicted urchin 80–85kDa iPLA2 is not present in the Uniprot database, the sequence was downloaded from PubMed. Human PLA2 sequences used for the analyses were sPLA2–P14555 and iPLA2–O60733 and murine PLA2 sequences were sPLA2–Q9WVF6 and iPLA2–P97819. Only those human and murine PLA2 sequences identified at the protein level were selected. As the predicted urchin sequences present in Uniprot or PubMed (66 sequences in total) do not contain the C2 domain characteristic of cPLA2, sequence alignment of cPLA2 isozymes was not carried out and there was no further testing for this isozyme. Building from the in-silico analysis, validation at the protein level was then carried out using a well-established top-down proteomic approach coupling 2DE with high-sensitivity western blotting using anti-human sPLA2 and iPLA2 antibodies to conserved regions [45,57,61].
2.2.4. Sample Fractionation for 2DE Western Blotting
After de-jellying and thorough washing, unfertilized urchin eggs were homogenized in intracellular media (IM; 220 mM K-glutamate, 500 mM glycine, 10 mM NaCl, 5 mM MgCl2, 5 mM EGTA, pH 6.7) supplemented with 2.5 mM ATP, 1 mM benzamidine HCl, 2 mg/mL aprotinin, 2 mg/mL pepstatin, 2 mg/mL leupeptin and 2 mM DTT) prior to standard isolation of high purity CSC or CV suspended in BIM [38,42,45,47,48,49,51,52]. For fractionation, CV were lysed using three volumes of ice-cold PIPES buffer (20 mM PIPES, pH 7.0 supplemented with protease, phosphatase and kinase inhibitors) for 90 s and isotonicity restored by adding an equal volume of 2 X IM (supplemented with protease, phosphatase and kinase inhibitors). Samples were centrifuged at 125,000× g, 4 °C, for 3 h to recover membrane pellets and CV luminal proteins in the supernatant [50]. Membranes were solubilized in 2DE buffer (4% CHAPS, 8 M Urea and 2 M Thiourea supplemented with protease, phosphatase and kinase inhibitors [50,57]) while supernatant containing CV luminal proteins was buffer exchanged with 3M urea solution using Amicon filters. Solubilized membrane proteins and concentrated CV luminal proteins were resolved using a well-established 2DE protocol involving isoelectric focusing (IEF) followed by SDS-PAGE in 1 mm thick 10% resolving gels [57]. Subsequent high sensitivity western blotting was essentially as previously described [41,42,45,61]. Briefly, resolved proteins were transferred to 0.2 µM pore size PVDF membrane (Bio-Rad) using buffer containing 25 mm Tris, 192 mm glycine, 20% methanol and 0.025% SDS at 90 Volts for 1 h. Blots were blocked for 1 h at room temperature (RT) in a solution containing 20 mM Tris, 150 mM NaCl, 0.1% Tween (TBST) and 5% non-fat dry milk or 20 mg/mL BSA. Blots were incubated overnight (at least 16 h) with constant rocking at 4 °C with anti-human sPLA2 (1:800) or iPLA2 (1:2000) primary antibodies. After washing, blots were incubated with secondary antibodies conjugated to horseradish peroxidase (HRP) for 2 h at RT and then detected using Luminata Crescendo western HRP substrate and the LAS 4000 Biomolecular Imager. Secondary antibody controls were always carried out in parallel western blots and parallel 2DE gels were stained with colloidal Coomassie Brilliant Blue (cCBB) [62,63].
2.2.5. Molecular Analyses to Detect CV Associated PLA2 Activities
Membrane and luminal fractions from purified CV suspensions were isolated using the same overall strategy as above except for the use of ice-cold BIM (pH 6.7) in the last step to maintain endogenous PLA2 activity. This also ensured tight coupling of functional (i.e., fusion) and molecular analyses. PLA2 activity in CV membrane and lumen fractions was measured by incubating 30 µg CV membrane or luminal protein with 200 picomoles NBD-PE or 170 picomoles NBD-PC for 30 s–10 min. PLA2 activity was then stopped by adding 0.2 µL HCl and all aliquots were immediately placed on ice for rapid isolation of total lipids; scaled-up experiments were carried out using more CV luminal protein (120 µg) and NBD-PE (664 picomoles) to best detect NBD-FFA species. An equal volume of BIM without CV luminal or membrane protein was used as a control to which the indicated amount of NBD-PC or -PE was added and incubated for 10 min.
To further establish whether catalytically active PLA2 was on the outer CV monolayer, well-established trypsin treatments were carried out to remove CV surface proteins and PLA2 activity was measured using the selective PLA2 substrate, PED6. Briefly, CV suspensions (2–4 mL; OD 0.95 + 0.04; n = 3) were treated with 700 U/mL trypsin for 1 h with gentle mixing every 15 min [42,45]. After treatment, CV suspensions were aliquoted into microplates and incubated with PED6 for 0–30 min; PLA2 activity was measured at 485 nm/520 nm (Ex/Em) using a POLARstar Omega microplate reader. Fusion assays were also carried out to confirm the fusion competence of the treated CV. To confirm removal of iPLA2 from the CV surface, western blotting was carried out on CV membrane proteins using the iPLA2 antibody. Protein isolation and SDS-PAGE were as above with the exception that SDS sample buffer was used for solubilizing CV membrane proteins and detection was with cCBB [41,42,45,62,63]. In parallel replicates, absorbance at A405 was measured regularly during the incubations to assess for possible CV lysis; any drastic decrease in absorbance indicated marked loss of CV due to bursting (as seen with the addition of ddH2O at the end of the experiment to confirm the intact state of CV).
2.2.6. Effects of PLA2 Activities on Fusion Assays
Standard fusion and docking/priming assays were carried out as previously described [42,43,45,47,48,49,50,51,52,54,56]. All experiments were carried out in BIM pH 6.7 supplemented with 2.5 mM ATP, 2 mM DTT and 1X protease inhibitors [41,42,43,45,51,52]. Briefly, CSC (0.5 mL, OD 0.56 ± 0.02, n = 3–14) and CV (OD 0.94 ± 0.03, n = 22) suspensions were treated with the indicated inhibitor concentrations. Immediately following treatments, suspensions were diluted with 3–5 volumes of ice-cold BIM, pH 6.7 and aliquoted into microplates. Each condition was challenged with increasing concentrations of [Ca2+]free in quadruplicate, consistent with established protocol and this was repeated in separate experiments as indicated (n) [42,43,45,47,48,49,50,51,52]. The decrease in OD representing CV fusion was measured using a POLARstar Omega microplate reader [53,54]. Final [Ca2+]free were measured with a Ca2+ sensitive electrode (Calcium Combination ISE, EDT directION Limited) as previously described [53,54]. All treatments were carried out at 25 ℃. Parallel solvent controls were carried out in every experiment. A separate aliquot in each experiment was also snap-frozen in liquid N2 and stored at −80 ℃ for subsequent molecular analyses.
3. Data Analyses
All data are reported as mean ± S.E.M. Two-sample two-tailed t-tests and 2-way ANOVA with Tukey’s multiple comparisons were performed using GraphPad PRISM 7 version 7.04 to assess the difference between experimental conditions (p ≤ 0.05 are considered significant).
4. Results
4.1. Identification of Endogenous PLA2 Activities on CSC and Their Effects on CSC Fusion
As CSC consist of CV endogenously docked on the PM and undergo Ca2+-triggered exocytosis in vitro, the potential presence and influence(s) of PLA2 activities was first assessed using CSC and selective PLA2 inhibitors. Treating CSC with LY311727 and BEL caused significant concentration-dependent decreases in endogenous FFA levels relative to a basal FFA level of 267.8 ± 29.8 femtomoles per µg CSC membrane protein (fmoles/μg MP) (Figure 1A; n = 3); 200 µM LY311727 reduced FFA levels to 122.7 ± 20.13 fmoles/μg MP while 100 µM and 500 µM BEL treatments reduced FFA levels below background values. No significant change in FFA levels was observed in any of the FKGK-11 treatments. As the lipid detection approach used is somewhat selective for unsaturated lipid species [56], in order to ensure a full assessment of potential PLA2 activities, CSC were labelled with NBD-PC (fluorophore at the sn-2 fatty acid); as previously documented, such exogenous substrates are readily incorporated into CSC and CV membranes [49]. The generation of 3.8 ± 1.2 picomoles NBD-FFA/μg MP from hydrolysis of the incorporated NBD-PC further confirmed endogenous PLA2 activity (Figure 1B; n = 3). The NBD-FFA was also generated from NBD-PE labelled CSC and CV (n = 1 confirmatory experiment, not shown). However, in contrast to Figure 1A, treating NBD-PC labelled CSC with increasing doses of BEL resulted in a concentration-dependent increase in NBD-FFA to 7.2 ± 3.0 and 9.8 ± 2.9 picomoles/μg MP. Similarly, a concentration-dependent increase in NBD-FFA to 8.9 ± 6.4 and 11.5 ± 8.2 picomoles/μg MP was observed in NBD-PC labelled CSC treated with 50 µM and 200 µM FKGK-11, respectively. No significant change in NBD-FFA levels were observed in any of the LY311727 treatments. As PLD also acts on PC, generation of 2.8 ± 0.5 picomoles NBD-PA/μg MP confirmed endogenous PLD activity (Figure 1B) consistent with previous findings [49]. However, none of the inhibitors caused significant change in NBD-PA with respect to the control. Importantly, the decrease in endogenous FFA levels (Figure 1A) and an increase in endogenous TAG, the de novo metabolite ([50,56]; Supplementary Figure 1A and also see [34,64,65]) in the BEL treated unlabelled CSC indicated the presence of a BEL sensitive PLA2 on CSC. However, the significant increase in NBD-FFA levels in BEL and FKGK-11 treated NBD-PC labelled CSC also indicated the presence of a BEL insensitive PLA2 species (Figure 1B). As BEL and FKGK-11 inhibit iPLA2 and LY311727 is a selective inhibitor of sPLA2 [26,35,64,66,67,68], the data suggested the presence of different catalytically active endogenous PLA2 in CSC preparations.
With the evidence of active endogenous PLA2, the effects of selective PLA2 inhibitors were then tested on CV-PM fusion. The native, docked CV-PM preparations exposed to increasing [Ca2+]free undergo exocytosis in vitro yielding a classic sigmoidal Ca2+ activity curve with an EC50 of 38.9 ± 6.4 µM [Ca2+]free (Figure 2; n = 14) [42,45,48,49,52,69,70]. Treating CSC with 20 µM and 200 µM LY311727 [66,68] significantly decreased fusion extent to 44.1 ± 5.3% and 33.6 ± 20.8%, respectively (Figure 2; n = 3–4). On treating CSC with 100 µM and 500 µM BEL [35,64,71,72] a significant decrease in the fusion extent to 53.7 ± 24.6% and 54.2 ± 7.9%, respectively, was observed; 500 µM BEL also caused a progressive rightward shift in Ca2+ sensitivity to an EC50 of 230.6 ± 101.5 µM [Ca2+]free (Figure 2; n = 3). CSC treated with 50 µM and 200 µM FKGK-11, another iPLA2 inhibitor [67], also significantly reduced fusion extent to 71.4 ± 28.5% and 31.3 ± 23.6%, respectively (Figure 2; n = 3). Although, LY311727, BEL and FKGK-11 inhibited endogenous FFA production and blocked triggered fusion; PLA2 species inhibited by LY311727 seem separate from the species which was inhibited by BEL and FKGK-11, in particular with reference to NBD-FFA generated from exogenous NBD-PC.
4.2. In-Silico Analysis
With initial evidence indicating the likely presence of more than one PLA2 isozyme on CSC and their potential role in regulating late steps of regulated exocytosis, we sought to identify these isozymes, beginning with sequence alignment to identify regions of homology in putative urchin PLA2 and characterized human and murine forms. The sequence alignment of sPLA2 and iPLA2 in the selected species clearly indicated conserved amino acid residues throughout the primary amino acid sequences, consistent with a high overall conservation from urchin to human (Figure 3) [40,73,74]. The urchin sPLA2 and iPLA2 forms showed high identity and similarity with both the human and murine species, particularly around the catalytic site histidine with >75% identity and similarity, as well as around the catalytic site serine with 100% identity and similarity (Figure 3A,B and Table 1). The sPLA2 also have a histidine in the Ca2+ binding loop with a XCGXGG consensus sequence to which LY311727 binds to inhibit the enzyme [66,68]. The putative urchin iPLA2 also contains a nucleotide binding site with a characteristic GGGVKG consensus sequence [35,75] and a caspase catalytic site with the DVTD consensus sequence [35,76,77]. Cysteine residues at C-67, C-185, C-240, C-344, C-465, C-550 and C-821 in the putative urchin iPLA2 are also conserved and are thus likely susceptible to alkylation during BEL treatments [78].
4.3. Western Blotting and Assays to Confirm Identity and Localization of PLA2 Isozymes
After identifying conserved catalytic and regulatory domains in the putative urchin isozymes (Figure 3 and Table 1), enzyme localization in CSC and CV was probed using selective antibodies. The anti-human sPLA2 antibody was raised against the entire length of human sPLA2 and would thus identify putative urchin sPLA2 considering the high identify and similarity, particularly within the catalytic region (Figure 3A and Table 1). Similarly, the anti-human iPLA2 recognizes a region between amino acid 399–704, which is also highly conserved in the putative urchin iPLA2 isozyme (54% identify, 67% similarity); this region also carries highly conserved GXSXG and GGGVKG catalytic and nucleotide binding consensus sequences (Figure 3B and Table 1). Western blotting of 2D gels identified two immune positive spots of ~14 kDa and pI of ~4.0 and ~4.9 in the CV lumen fraction using the sPLA2 antibody (Figure 4A). An immune positive spot of ~63 kDa and pI ~6.2 and two immune positive spots of ~63 kDa and pI ~5.2 and 5.4 were detected in the CSC and CV membrane fractions, respectively, using the iPLA2 antibody (Figure 4B,C, respectively). Of note is the relative high abundance of iPLA2 on the CSC membrane in comparison to the CV membrane.
To further and more specifically localize the PLA2 isozymes and explore possible substrate preferences, NBD-PE and -PC were used to assess the CV associated enzyme activities. First, an isolate of CV lumen proteins was incubated with NBD-PE or -PC for different interval (Figure 5A; n = 3). The NBD-PE in controls after 10 min incubation was 12.3 ± 0.9 picomoles which was significantly reduced to 9.0 ± 1.2, 7.4 ± 1.2, 6.8 ± 1.0 and 3.8 ± 2.3 picomoles following 30 s, 2 min, 5 min and 10 min incubations, respectively (Figure 5A). The generation of NBD-FFA having a retention factor (Rf) similar to NBD-ARA indicated PLA2 activity; an ~70–90% increase in NBD-FFA over time was observed (Figure 5A; n = 2–3). Notably, a very large increase in NBD-FFA in CV aliquots that were pre-incubated with 104.0 µM [Ca2+]free confirmed Ca2+ promoted CV luminal PLA2 activity (Figure 5B, n = 3). In contrast, 18.4 ± 2.0 picomoles NBD-PC was detected in buffer controls and no significant change was detected even after 10 min incubation with the CV luminal protein (Figure 5A). This correlated with no detection of NBD-FFA species in NBD-PC labelled CSC (n = 2 confirmatory experiments, not shown). Thus, a decrease in NBD-PE and parallel increase in NBD-FFA over time and a large increase in the generation of NBD-FFA in the presence of higher [Ca2+]free confirmed the presence of a Ca2+ dependent catalytically active sPLA2 in the isolated CV luminal fraction, which is selective for PE over PC.
The substrate preference of the ~63kDa CV membrane iPLA2 was also investigated by incubating isolated CV membrane fragments with NBD-PE or -PC (Figure 6A; n = 2–3). Buffer without CV membrane was used as the control to which the indicated amount of NBD-PC or -PE was added and incubated for 10 min. In all incubations, no significant change in the NBD-labelled lipids nor generation of NBD-FFA was observed. Therefore, to confirm whether the apparent membrane associated iPLA2 was lost or became inactive during CV membrane isolation, intact CV were assayed for PLA2 activity following trypsin treatment and loss of iPLA2 was confirmed by western blotting. Loss of the ~63 kDa iPLA2 protein band following trypsin treatment confirmed the presence of iPLA2 on the CV surface (Figure 6B, n = 3; see also Figure S2). Both control and trypsin treated CV were then incubated with the PLA2 selective substrate PED6—a glycerophosphoethanolamine with a BODIPY dye-labelled sn-2 acyl chain and a dinitrophenyl quencher-modified head group [79,80]. The cleavage of the BODIPY dye from the sn-2 position by the action of PLA2 resulted in decreased quenching by the head group which specifically indicated PLA2 activity. Relative to the 0 min control, there was no detectable PLA2 activity in the trypsin treated CV, until 2 min. No significant change in the fluorescence intensity between 2–30 min in trypsin treated CV, relative to 0 min, indicated a complete loss of iPLA2 activity that correlated with loss of the iPLA2 band on the western blots (Figure 6C; n = 3). Absorbance measurements of parallel samples confirmed that there was no major loss of CV over the course of these incubations (not shown).
Considering the apparent complete loss of the ~63 kDa iPLA2 from the external membrane of intact CV following trypsin treatment and the corresponding loss of activity assessed using the selective PED6 substrate (Figure 6B,C), we also tested these same batches of control and trypsin treated CV for functional activity. Parallel standard and settle fusion assays were thus carried out (Figure 6D,E, n = 2–3). In the standard assay, there was no significant change in the fusion extent although Ca2+ sensitivity appeared right-shifted following trypsin treatment; however, the shift was not significant relative to the untreated controls. Notably, in the settle assay used to assess docking and priming [42,47,49,50,51,52,54], fusion extent was significantly decreased to 54.3 ± 7.2% following trypsin treatment (Figure 6E; n = 3, (p < 0.004)).
4.4. The Effect of PLA2 Activities on CV-CV Fusion
As in previous work, to enable a tight focus on the late steps of Ca2+ -triggered exocytosis, including the fusion mechanism itself, without having to contend with the ‘background’ of the PM, high purity CV preparations were used to further assess the potential influences of the endogenous CV-associated PLA2 activities. Both the sPLA2 and iPLA2 were thus again targeted with selective inhibitors to better assess their potential influence(s) on the late steps of regulated exocytosis (Figure 7A,B; n = 3–19). The standard endpoint CV-CV fusion assays yielded a characteristic sigmoidal Ca2+ activity curve with an EC50 of 44.8 ± 3.8 µM [Ca2+]free (Figure 7A; n = 19), which was translationally invariant with CV-PM fusion (Figure 1; n = 14) [42,45,49,51]. A significant decrease in fusion extent to 95.3 ± 2.9% and 80.0 ± 10.7%, was observed in CV treated with 20 µM and 200 µM LY311727, respectively (Figure 7A). Fusion of CV treated with 100 µM and 500 µM BEL were also reduced to 96.0 ± 3.2% and 90.0 ± 6.4%, respectively (Figure 7A); following treatment with 50 µM and 200 µM FKGK-11 fusion extent was reduced to 87.4 ± 8.5% and 75.4 ± 7.3%, respectively (Figure 7A). A significant right-shift in Ca2+ sensitivity, to yield EC50 of 78.9 ± 9.4 µM [Ca2+]free and 79.8 ± 7.5 µM [Ca2+]free was also observed following treatment with 50 µM and 200 µM FKGK-11.
When tested in the settle assay, CV treated with all doses of the inhibitors showed further decreases in fusion extent. These concentration-dependent decreases in fusion were to 89.9 ± 4.6% and 53.5 ± 13.4% in 20 µM and 200 µM LY311727 treated CV, respectively; 91.3 ± 4.6% and 87.6 ± 6.6% in 100 µM and 500 µM BEL treated CV, respectively; and 77.0 ± 16.0% and 58.9 ± 10.9% in 50 µM and 200 µM FKGK-11 treated CV, respectively, confirming the role(s) of PLA2 isozymes in promoting and maintaining CV priming and/or docking (Figure 7B). To confirm that changes in the CV-CV fusion parameters were associated with perturbed PLA2 activities, total lipid changes were assessed from the same preparations following inhibitor treatments. Treating CV with LY311727, BEL and FKGK-11 caused significant concentration-dependent decreases in endogenous FFA levels, relative to a basal FFA level of 15.7 ± 0.51 fmoles/μg MP (Figure 8; n = 3–4). FFA levels decreased to 12.9 ± 1.3 and 8.9 ± 3.5 fmoles/μg MP in CV treated with 20 µM and 200 µM LY311727, respectively. Additionally, a significant increase in the endogenous PE levels in the CV treated with 200 µM LY311727 (Table 2), confirmed the inhibitory action of LY311727 on PE-selective PLA2. Treating CV with 500 µM BEL also significantly reduced FFA levels to 1.3 ± 6.7 fmoles/μg MP (Figure 8A,B; n = 4–12) and increased TAG levels to 376.7 ± 22.5 picomoles/μg MP, relative to the basal level of 217.2 ± 40.9 picomoles in the control (Figure S1B, n = 3–8; also seen in the BEL treated CSC, Figure S1A, n = 3–5).
5. Discussion
Using bioinformatics and a top-down proteomic approach combined with lipid analyses of isolated native CSC and CV, here we have identified endogenous, vesicle associated PLA2—a sPLA2 in the CV lumen and an iPLA2 on the CV surface—and also demonstrated that their activities play a role in promoting and maintaining the docked and/or primed state of secretory vesicles. The results are consistent with previous findings in platelets that PLA2 activity is not essential for fusion per se but has clear modulatory roles late in the exocytotic pathway [81].
5.1. PLA2 Activity and Their Effects on CSC Fusion
Initial assessments of CSC confirmed the existence of endogenous PLA2 activities and that these were associated with the late steps of the regulated exocytotic pathway (Figure 1 and Figure 2). Blockade of these PLA2 activities using the well-characterized iPLA2 inhibitors, BEL [35,64,66,78] and FKGK-11 [35,67] and the sPLA2 inhibitor LY311727 [68], caused significant decreases in endogenous FFA levels (Figure 1A) and an increase in PE levels (Table 2). Importantly, a significant decrease in endogenous FFA but increase in NBD-FFA in BEL treated CSC also indicated the presence of BEL sensitive as well as insensitive PLA2 isozymes, the latter seemingly an sPLA2 (Figure 1A,B). Furthermore, to assess for potential nonspecific inhibitory effects of BEL on other enzymes [78], unlabelled and NBD-PC labelled CSC were treated with another iPLA2 inhibitor, FKGK-11. The significant concentration-dependent increase in NBD-FFA (similar to that seen with BEL; Figure 1B) supported the interpretation that BEL and FKGK-11 targeted at least one common molecular entity. This might also indicate that alternate iPLA2 isoforms or proteoforms compensated for the loss, in the presence of BEL or FKGK-11. In contrast, using LY311727 to block sPLA2 resulted in a concentration-dependent decrease in endogenous FFA with no change in NBD-FFA, generated from NBD-PC labelled CSC (or NBD-PE labelled CSC; n = 1, not shown). These data indicated an active sPLA2 that could not access the exogenous NBD-PE or -PC (Figure 1A,B), despite the fact that NBD-PE was also seen to be a preferred substrate of CV luminal PLA2 (Figure 5A). Also, no significant change in the fluorescence intensity in the 30 min control, relative to the 0 min control (Figure 6C), indicated that 30 min was insufficient for any putative CV flippase to translocate PED6 (a modified PE) toward the CV lumen; had there been such translocation of PED6, a significant increase in fluorescence intensity should have occurred within 30 min due to luminal sPLA2 activity.
Notably, a significant increase in TAG in the BEL treated unlabelled CSC provided an early indication that iPLA2 could be the isozyme on CSC (Figure S1A and [34,64]). The proposed ‘housekeeping’ function of iPLA2 is to generate lyso-phospholipid acceptors for the incorporation of FFA into membrane phospholipids [34,35,64,82]. Upon iPLA2 inhibition, lyso-phospholipid acceptor levels decrease, consequently leading to an increase in FFA concentration [29,64]. Under such a condition, the bulk of FFA is incorporated in the de novo metabolites TAG, DAG and MAG (Figure S1A,B) [64]. BEL also inhibits magnesium-dependent phosphatidate phosphohydrolase (PAP), an enzyme catalysing the conversion of PA to DAG; however, there were no changes in PA in BEL treated CSC (Figure 1B).
Overall, the data suggested the presence of more than one catalytically active form of PLA2 on CSC, some of which may be strategically localized to maintain the docked and release-ready state of CV on the PM [29]. Blocking PLA2 activities using well characterized iPLA2 inhibitors (BEL [78] and FKGK-11 [35,67]) and an sPLA2 inhibitor (LY311727 [68]) caused significant inhibition of CSC fusion suggesting that even fully docked and fusion-ready CV are sensitive to changes in local PLA2 activity (Figure 1B and Figure 2).
5.2. Bioinformatics
With functional confirmation of the presence of active PLA2 species and association of their metabolites with the late steps of Ca2+-triggered exocytosis, we used a combined bioinformatics and top-down proteomics approach to identify and localize the active isoforms. Orthologs and paralogs are hypothesized to share high structural and functional similarity [83]; proteins essential to fundamental molecular mechanisms have a similar structure and function from urchin to mammals [37,73,74,83]. Accordingly, there was substantial homology in amino acid sequence between urchin and mammalian sPLA2 and iPLA2, particularly at the catalytic sites (Figure 3 and Table 1) [37]. The putative urchin sPLA2 shares conserved catalytic and regulatory regions of >75% identity and similarity with the mammalian orthologs (Table 1), including the highly conserved histidine and aspartic acid in the catalytic site consensus sequence (HDCCY) and the histidine in the Ca2+ binding loop (HCGVGG) (Figure 3A), at which LY311727 binds via its amide group [66]. Similarly, the iPLA2 share conserved catalytic and regulatory domains, including the conserved active site serine in the GXSXG consensus sequence that interacts with FKGK-11 [37,69] and the conserved cysteine residues are alkylated by the active BEL metabolites [78]. The urchin iPLA2 thus shares conserved catalytic and regulatory domains with the human and murine orthologs (Figure 3B and Table 1). Additionally, it also contains an ATP binding consensus motif (GGGVKG) and a N-terminal caspase-3 cleavage site (DVTD), that are all characteristic of iPLA2β (Figure 3) [35,84]. Notably, iPLA2β and sPLA2 genes are conserved in lower eukaryotes, further indicating an essential role of these enzymes [37].
5.3. Top-Down Proteomics Identify CV Associated PLA2 Isozymes
Based on the bioinformatics we then carried out targeted top-down proteomic analyses using 2DE and immunoblotting. These analyses identified a highly conserved catalytically active ~14 kDa CV luminal sPLA2 isozyme (Figure 4A) [25,26,36,85,86] having substrate preference for PE over PC (Figure 5A,B) [26]. Indeed, there are other reports of 14 kDa sPLA2 species having a substrate selectivity for PE over PC [17,87]. An increase in the activity of this CV luminal sPLA2 species in the presence of [Ca2+]free was also confirmatory of its identification (Figure 5B) [25,27]. Similarly, using an antibody to the human form, we detected an ~63 kDa iPLA2 on both CSC and CV membranes (Figure 4B,C), although having different isoelectric points (pI). One of the most common post-translation modifications (PTM) causing a shift in pI is phosphorylation, resulting in an increase in the net negative charge on the protein. Hence, CV membrane iPLA2 isozymes with pI of ~5.2 and 5.4 may be phosphorylated (or otherwise modified) species relative to the bulk of the enzyme found on CSC (pI of ~6.2) (Figure 4B,C); the broad potential influence of protein phosphorylation on the late steps of exocytosis has been well documented [52]. The ~63kDa iPLA2 is proposed to arise from the proteolytic cleavage of a nascent full length 80–85 kDa iPLA2 [35,76,77]. Therefore, together with in-silico analysis (Figure 3B), identification of an ~63 kDa iPLA2 on the CV membrane (Figure 4C) and the significant decrease in endogenous FFA in BEL and FKGK-11 treated CV (Figure 8A,B) with a parallel increase in TAG (Figure S1B), confirmed a catalytically active membrane bound CV iPLA2 [35,64,76,77,82]. Notably, on comparing CSC and CV data (Figure 4B,C), it would seem that much more of the iPLA2 may be localized to the PM, some (small) fraction presumably near or at sites of CV docking and fusion. The putative urchin iPLA2 contains 57 potential trypsin cleavage sites, predicted by PeptideMass [88] and thus cleavage at even a few of these sites would destroy the enzyme. Hence, loss of the 63 kDa CV iPLA2 following trypsin treatment (Figure 6B) and associated decrease in signal from the PLA2 selective PED6 substrate (Figure 6C), further confirmed the presence of an active iPLA2 on the external CV membrane. Thus, proteolytic removal of CV surface proteins, including iPLA2, caused a significant decrease in fusion extent to 54.3 ± 7.2% in the settle fusion assay that evaluates the priming and docking capacity of CV (Figure 6E), indicating that vesicle surface proteins including iPLA2 and SNAREs [29,41,42,43,44,45,89]―and possibly other as yet unidentified proteins―are critical for priming and/or docking. This is consistent with a suggested link between the CV surface PLA2 activity and influences on the late steps of the exocytotic pathway [29].
5.4. CV Associated PLA2 Isozymes Regulate Docking and/or Priming
Finally, we sought to test whether the identified PLA2 isozymes are components of the minimal molecular machinery present on CV which enables docking, Ca2+ sensing and membrane merger [38,42,43,45,47,48,49,50,51,52,55,90,91,92]. Thus, CV were treated with selective PLA2 inhibitors and changes in the fusion parameters were assessed using the well-established standard and settle fusion assays [38,42,43,45,47,48,49,50,51,52,53,54,90,91,92]. Consistent with the significant concentration-dependent decrease in fusion extent in CSC fusion assays, comparable effects were seen in the standard CV fusion assay following treatments with LY311727, BEL and FKGK-11 (Figure 7A). However, a far more pronounced decrease in the fusion extent in the settle assays (Figure 7B) confirmed that CV associated PLA2 isozymes or their metabolites play a critical role in docking and/or priming. The significant concentration-dependent decrease in endogenous CV FFA following the inhibitor treatments (Figure 8A,B), significant increase in PE (Table 2; in CV treated with 200 µM LY311727) and parallel increase in TAG (Figure S1B; in CV treated with 500 µM BEL) confirmed that these inhibitors targeted CV associated PLA2 isozymes.
Furthermore, iPLA2 hydrolyse BEL to generate diffusible Bromomethyl Keto Acid (BMKA), which alkylates cysteine thiols in iPLA2 causing its inhibition, in addition to non-specific alkylation of cysteine thiols present in other proteins [78]. Therefore, the significant reduction in the Ca2+ sensitivity of CSC treated with 500 µM BEL (Figure 2), relative to a lack of effect on CV treated identically (Figure 7A,B), suggested that BMKA acted (to some extent) like N-ethylmaleimide (NEM) in CSC [90,92]. This indicates additional thiol sites on the PM that regulate efficiency of the fusion mechanism (i.e., Ca2+ sensing). In terms of targeted inhibition, it should be noted that the compounds, LY311727, BMKA and FKGK-11 are highly hydrophobic with log P values of 1.92 ± 0.62, 3.90 ± 0.37 and 5.40 ± 0.89, respectively, where the partition coefficient (P) = [organic]/[aqueous] (calculated using ADC/ChemSketch [93]). Hence, these compounds have a high likelihood to intercalate into or cross the CV membrane. Indeed, the significant concentration-dependent decrease in fusion extent upon 200 µM LY311727 treatments, to 33.6 ± 20.8%, 80.0 ± 10.7% and 53.5 ± 13.4% in the CSC, CV-CV standard and settle fusion assays, respectively, (Figure 2A,B and Figure 7A,B) and; the parallel decrease in endogenous FFA (Figure 1A and Figure 8A,B) indicated that inhibition of the CV luminal sPLA2 reduced CV priming and/or docking. Although BMKA and FKGK-11 may also have crossed the CV membrane, it seems unlikely that these would inhibit the sPLA2―its sulfhydryl groups would already be engaged in intra-molecular disulphide bonding and thus would not be susceptible to BMKA induced alkylation [78] and the absence of a catalytic site serine would obviate FKGK-11 binding [67]. Therefore, overall, the inhibitors chosen are expected to be highly selective for the PLA2 species in question and thus reflect selective influences on the functional assays.
5.5. Concluding Remarks
According to the Stalk-pore hypothesis, the fusion of two distinct membrane bilayers proceeds through sequential steps resulting in the localized mixing of proximal monolayers followed by fusion of the distal monolayers [94,95,96,97]. This enables vesicle content mixing (homotypic fusion) or release (heterotypic fusion) and subsequent content dispersal. PLA2 activity produces both negative and positive curvature lipids—LPC and FFA, respectively—which are known to inhibit or promote fusion depending on their site of incorporation [10,11,29,94,95,98]. In this regard, localization of the endogenous catalytically active PLA2 isozymes that supply LPC and FFA near the docking and/or fusion site is of critical importance. The internal CV microenvironment has a pH of ~5.5 and contains ~100 mM calcium, mostly in a bound state, as has also been found in other (mammalian) secretory vesicles [30,86,99,100,101]; however, the estimated global luminal [Ca2+]free is ~1–10 µM, occurring as transients, linked in part to the opening of p-type Ca2+ channels [30]. As these are global measures, the local [Ca2+]free at the CV membrane would be much higher. This is also consistent with the estimates of luminal [Ca2+]free in chromaffin granules under basal conditions [100]. Hence, in response to CV luminal Ca2+ transients in vivo, the activity of localized luminal sPLA2 would increase, preferentially hydrolysing PE on the inner CV monolayer (Figure 4A and Figure 5A,B ), at or immediately adjacent to the docking/fusion site. Thus, under basal conditions, the external CV membrane associated iPLA2 generates LPC and FFA by cleaving PC on the outer CV monolayer, while luminal sPLA2 produces LPE and FFA on the inner CV monolayer. Such localization of iPLA2 on the outer vesicle (and inner plasma) membrane, near docking/fusion sites, would block spontaneous merger of the vesicle and PM due to high local levels of LPC at membrane contact sites, in particular considering the substantially higher positive intrinsic curvature of LPC relative to LPE [98]. As such, this might act as a native fusion ‘brake’ that must be overcome by the Ca2+-triggered fusion mechanism [29], in which basal LPC production is somewhat balanced by intermittent LPE production via transient sPLA2 activity (Figure 9). In this regard and that of FFA (see below) it would also be important to assess the distribution of fatty acid species at sn-1 and sn-2 of the endogenous CV membrane PC and PE. The net accumulation of FFA on cis or trans monolayers and any associated effects would be dependent on the rate of passive diffusion, flipping efficiency, rate of metabolism, site of incorporation, chain length and degree of unsaturation [10,95,102,103,104,105,106,107]. Notably, perhaps complimentary to the actions of LPC and LPE, PLA2 derived FFA has been proposed to support the formation of trans SNARE complexes to maintain the primed and/or docked state of vesicles [4,29,108,109]. Importantly, basal CV luminal sPLA2 activity supported CV-PM and CV-CV fusion (Figure 2, Figure 6D,E and Figure 7A,B); whether the activity is also sensitive to transient changes in luminal [Ca2+]free and how these may be linked to the final membrane merger steps are unknown. However, the likely coordinated action (i.e., cross-talk) of the CV luminal sPLA2 and outer membrane bound iPLA2 in maintaining the docked, fusion-ready state of vesicles on the PM is evident as both interventions used here―pharmacological inhibition of CV luminal sPLA2 and membrane associated iPLA2 and proteolytic removal of iPLA2―significantly impaired priming and/or docking.
6. Conclusions
The data confirm (i) the presence of endogenous, catalytically active CV associated PLA2 isozymes―a CV luminal sPLA2 and an iPLA2 associated with the outer CV membrane; and (ii) that proteolytic removal of CV surface proteins including iPLA2 or the pharmacological inhibition of both the isozymes significantly reduces priming and/or docking. Based on this as well as previously measured vesicular Ca2+ transients, basal Ca2+ dependent CV luminal sPLA2 activity and CV membrane attached iPLA2 would appear to act together to support efficient vesicle priming and/or docking. While intriguing, this warrants further study, particularly relating to the apparent effects of FFA on the formation of SNARE complexes and/or in modulating membrane biophysical properties, as FFA is also known to induce the transition from the bilayer to the hexagonal phase upon its incorporation in the membrane [10,107,110].
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4409/8/4/303/s1. Figure S1: Endogenous TAG levels in (A) CSC; n = 3–5 and (B) CV; n = 3–8 treated with the indicated doses of LY311727, BEL and FKGK-11, including representative chromatograms showing changes in neutral lipids. Figure S2: (A) SDS-PAGE of total CV membrane protein isolated from trypsin treated CV and the parallel untreated control; (B) secondary antibody control western blot to identify non-specific protein binding. A total of 5 µg CV membrane protein per lane was resolved.
Author Contributions
J.R.C. conceptualized the study and provided all resources and funding. J.R.C. and D.D. designed the methodology. D.D. carried out the investigation, data curation and analysis. D.D. and J.R.C. interpreted the data. D.D. wrote the original draft. D.D. and J.R.C. wrote the final manuscript.
Funding
This study is supported by funding from the National Health and Medical Research Council (NHMRC–APP1065328) of Australia and an anonymous family foundation.
Acknowledgments
J.R.C. is grateful to an anonymous family foundation for providing initial financial support for this work and acknowledges targeted funding from the National Health and Medical Research Council (NHMRC–APP1065328). DD thanks Western Sydney University (WSU), School of Medicine (SOM) for a Post Graduate Research Award. DD and JRC thank Ashleigh Deschamps WSU, SOM for urchin collections and maintenance of the aquatic facility. We also thank David Messiah, Lana Kajlich and the Underwater Research Group of NSW for additional urchin collections.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ARA | arachidonic acid |
| [Ca2+]free | free calcium ion concentration |
| cCBB | colloidal Coomassie brilliant blue G-250 dye |
| CHAPS | 3-[(3-Cholamidopropyl) dimethylammonio]-1-propanesulfonate hydrate |
| CHOL | cholesterol |
| CE | cholesterol ester |
| CSC | cell surface complexes |
| CV | cortical vesicles |
| FFA | free fatty acid |
| DAG | diacylglycerol |
| LPC | lyso-phosphatidylcholine |
| PC, PE, PI, PG, PS | phosphatidyl-choline, -ethanolamine, -inositol, -glycerol, -serine |
| MAG | monoacylglycerol |
| pI | isoelectric point |
| PED6 | (N-((6-(2,4-dinitrophenyl) amino) hexanoyl)-2-(4,4-difluoro-5,7-dimethyl-4-Bora-3a,4a-diaza-s-Indacene-3-pentanoyl)-1-hexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt |
| SV | synaptic vesicles |
| TAG | triacylglycerol |
References
- Frye, R.A.; Holz, R.W. The relationship between arachidonic acid release and catecholamine secretion from cultured bovine adrenal chromaffin cells. J. Neurochem. 1984, 43, 146–150. [Google Scholar] [CrossRef]
- Frye, R.A.; Holz, R.W. Arachidonic acid release and catecholamine secretion from digitonin-treated chromaffin cells: Effects of micromolar calcium, phorbol ester and protein alkylating agents. J. Neurochem. 1985, 44, 265–273. [Google Scholar] [CrossRef]
- Juhl, K.; Hoy, M.; Olsen, H.L.; Bokvist, K.; Efanov, A.M.; Hoffmann, E.K.; Gromada, J. cPLA2 alpha-evoked formation of arachidonic acid and lysophospholipids is required for exocytosis in mouse pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E73–E81. [Google Scholar] [CrossRef]
- Latham, C.F.; Osborne, S.L.; Cryle, M.J.; Meunier, F.A. Arachidonic acid potentiates exocytosis and allows neuronal SNARE complex to interact with Munc18a. J. Neurochem. 2007, 100, 1543–1554. [Google Scholar] [CrossRef]
- Matsuzawa, A.; Murakami, M.; Atsumi, G.; Imai, K.; Prados, P.; Inoue, K.; Kudo, I. Release of secretory phospholipase A2 from rat neuronal cells and its possible function in the regulation of catecholamine secretion. Biochem. J. 1996, 318, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, N.; Schook, W.; Puszkin, S. Interaction of brain synaptic vesicles induced by endogenous Ca2+ -dependent phospholipase A2. Science 1982, 216, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.; Berman, J.D.; Middleton, W.; Brendle, J. Botulinum toxin inhibits arachidonic acid release associated with acetylcholine release from PC12 cells. J. Biol. Chem. 1993, 268, 11057–11064. [Google Scholar] [PubMed]
- Ray, P.; Ishida, H.; Millard, C.B.; Petrali, J.P.; Ray, R. Phospholipaise A2 and arachidonic acid-mediated mechanism of neuroexocytosis: A possible target of botidinum neurotoxin A other then SNAP-25. J. Appl. Toxicol. 1999, 19, S27–S28. [Google Scholar] [CrossRef]
- Karli, U.O.; Schafer, T.; Burger, M.M. Fusion of neurotransmitter vesicles with target membrane is calcium independent in a cell-free system. Proc. Natl. Acad. Sci. USA 1990, 87, 5912–5915. [Google Scholar] [CrossRef] [PubMed]
- Chernomordik, L.; Chanturiya, A.; Green, J.; Zimmerberg, J. The hemifusion intermediate and its conversion to complete fusion: Regulation by membrane composition. Biophys. J. 1995, 69, 922–929. [Google Scholar] [CrossRef]
- Chernomordik, L.V.; Vogel, S.S.; Sokoloff, A.; Onaran, H.O.; Leikina, E.A.; Zimmerberg, J. Lysolipids reversibly inhibit Ca2+-, GTP- and pH-dependent fusion of biological membranes. FEBS Lett. 1993, 318, 71–76. [Google Scholar] [CrossRef]
- Reese, C.; Mayer, A. Transition from hemifusion to pore opening is rate limiting for vacuole membrane fusion. J. Cell Biol. 2005, 171, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Uriarte, S.M.; Powell, D.W.; Luerman, G.C.; Merchant, M.L.; Cummins, T.D.; Jog, N.R.; Ward, R.A.; McLeish, K.R. Comparison of Proteins Expressed on Secretory Vesicle Membranes and Plasma Membranes of Human Neutrophils. J. Immunol. 2008, 180, 5575–5581. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.L.; Bark, S.J.; Funkelstein, L.; Mosier, C.; Yap, A.; Kazemi-Esfarjani, P.; La Spada, A.R.; Sigurdson, C.; O’Connor, D.T.; Hook, V. Proteomics of Dense Core Secretory Vesicles Reveal Distinct Protein Categories for Secretion of Neuroeffectors for Cell−Cell Communication. J. Proteome Res. 2010, 9, 5002–5024. [Google Scholar] [CrossRef]
- Brunner, Y.; Couté, Y.; Iezzi, M.; Foti, M.; Fukuda, M.; Hochstrasser, D.F.; Wollheim, C.B.; Sanchez, J.-C. Proteomics Analysis of Insulin Secretory Granules. Mol. Cell. Proteom. 2007, 6, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Schvartz, D.; Brunner, Y.; Coute, Y.; Foti, M.; Wollheim, C.B.; Sanchez, J.C. Improved characterization of the insulin secretory granule proteomes. J. Proteom. 2012, 75, 4620–4631. [Google Scholar] [CrossRef] [PubMed]
- Chock, S.P.; Schmauder-Chock, E.A.; Cordella-Miele, E.; Miele, L.; Mukherjee, A.B. The localization of phospholipase A2 in the secretory granule. Biochem. J. 1994, 300, 619–622. [Google Scholar] [CrossRef]
- Bingham, C.O.; Fijneman, R.J.A.; Friend, D.S.; Goddeau, R.P.; Rogers, R.A.; Austen, K.F.; Arm, J.P. Low Molecular Weight Group IIA and Group V Phospholipase A2 Enzymes Have Different Intracellular Locations in Mouse Bone Marrow-derived Mast Cells. J. Biol. Chem. 1999, 274, 31476–31484. [Google Scholar] [CrossRef]
- Moskowitz, N.; Puszkin, S.; Schook, W. Characterization of brain synaptic vesicle phospholipase A2 activity and its modulation by calmodulin, prostaglandin E2, prostaglandin F2 alpha, cyclic AMP and ATP. J. Neurochem. 1983, 41, 1576–1586. [Google Scholar] [CrossRef]
- Moskowitz, N.; Schook, W.; Puszkin, S. Regulation of endogenous calcium-dependent synaptic membrane phospholipase A2. Brain Res. 1984, 290, 273–280. [Google Scholar] [CrossRef]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Grønborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brügger, B.; Ringler, P.; et al. Molecular Anatomy of a Trafficking Organelle. Cell 2006, 127, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Marco, M.; Jacqueline, B.; Carsten, C.; Michael, K.; Herbert, Z.; Walter, V. Immunoisolation of two synaptic vesicle pools from synaptosomes: A proteomics analysis. J. Neurochem. 2005, 95, 1732–1745. [Google Scholar] [CrossRef]
- Heo, S.; Diering, G.H.; Na, C.H.; Nirujogi, R.S.; Bachman, J.L.; Pandey, A.; Huganir, R.L. Identification of long-lived synaptic proteins by proteomic analysis of synaptosome protein turnover. Proc. Natl. Acad. Sci. USA 2018, 115, E3827–E3836. [Google Scholar] [CrossRef]
- Boyken, J.; Grønborg, M.; Riedel, D.; Urlaub, H.; Jahn, R.; Chua, J.J.E. Molecular Profiling of Synaptic Vesicle Docking Sites Reveals Novel Proteins but Few Differences between Glutamatergic and GABAergic Synapses. Neuron 2013, 78, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A. The growing phospholipase A2 superfamily of signal transduction enzymes. Trends Biochem. Sci. 1997, 22, 1–2. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Yang, H.C.; Rosenberger, T.A.; Horrocks, L.A. Phospholipase A2 and its role in brain tissue. J. Neurochem. 1997, 69, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Kudo, I.; Murakami, M.; Hara, S.; Inoue, K. Mammalian non-pancreatic phospholipases A2. Biochim. Biophys. Acta (Bba) Lipids Lipid Metab. 1993, 1170, 217–231. [Google Scholar] [CrossRef]
- Mounier, C.M.; Ghomashchi, F.; Lindsay, M.R.; James, S.; Singer, A.G.; Parton, R.G.; Gelb, M.H. Arachidonic Acid Release from Mammalian Cells Transfected with Human Groups IIA and X Secreted Phospholipase A2 Occurs Predominantly during the Secretory Process and with the Involvement of Cytosolic Phospholipase A2-α. J. Biol. Chem. 2004, 279, 25024–25038. [Google Scholar] [CrossRef]
- Dabral, D.; Coorssen, J.R. Phospholipase A2: Potential roles in native membrane fusion. Int. J. Biochem. Cell Biol. 2017, 85, 1–5. [Google Scholar] [CrossRef]
- Raveh, A.; Valitsky, M.; Shani, L.; Coorssen, J.R.; Blank, P.S.; Zimmerberg, J.; Rahamimoff, R. Observations of Calcium Dynamics in Cortical Secretory Vesicles. Cell Calcium 2012, 52, 217–225. [Google Scholar] [CrossRef]
- Estévez-Herrera, J.; Domínguez, N.; Pardo, M.R.; González-Santana, A.; Westhead, E.W.; Borges, R.; Machado, J.D. ATP: The crucial component of secretory vesicles. Proc. Natl. Acad. Sci. USA 2016, 113, E4098–E4106. [Google Scholar] [CrossRef]
- Winkler, H.; Westhead, E. Th molecular organization of adrenal chromaffin granules. Neuroscience 1980, 5, 1803–1823. [Google Scholar] [CrossRef]
- Jaime, S.; Laura, V.; Marcial, C.; Esther, H.S.; Fonteriz, R.I.; Lobaton, C.D.; Mayte, M.; Alfredo, M.; Javier, A. Calcium dynamics in bovine adrenal medulla chromaffin cell secretory granules. Eur. J. Neurosci. 2008, 28, 1265–1274. [Google Scholar] [CrossRef]
- Balsinde, J.; Balboa, M.A.; Dennis, E.A. Antisense inhibition of group VI Ca2+-independent phospholipase A2 blocks phospholipid fatty acid remodeling in murine P388D1 macrophages. J. Biol. Chem. 1997, 272, 29317–29321. [Google Scholar] [CrossRef] [PubMed]
- Ramanadham, S.; Ali, T.; Ashley, J.W.; Bone, R.N.; Hancock, W.D.; Lei, X. Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J. Lipid Res. 2015, 56, 1643–1668. [Google Scholar] [CrossRef]
- Six, D.A.; Dennis, E.A. The expanding superfamily of phospholipase A(2) enzymes: Classification and characterization. Biochim. Biophys. Acta 2000, 1488, 1–19. [Google Scholar] [CrossRef]
- Murakami, M.; Taketomi, Y.; Miki, Y.; Sato, H.; Hirabayashi, T.; Yamamoto, K. Recent progress in phospholipase A2 research: From cells to animals to humans. Prog. Lipid Res. 2011, 50, 152–192. [Google Scholar] [CrossRef]
- Abbineni, P.S.; Wright, E.P.; Rogasevskaia, T.P.; Killingsworth, M.C.; Malladi, C.S.; Coorssen, J.R. The Sea Urchin Egg and Cortical Vesicles as Model Systems to Dissect the Fast, Ca2+-Triggered Steps of Regulated Exocytosis. In Exocytosis Methods; Thorn, P., Ed.; Humana Press: Totowa, NJ, USA, 2014; pp. 221–241. [Google Scholar]
- Zimmerberg, J.; Coorssen, J.R.; Vogel, S.S.; Blank, P.S. Sea urchin egg preparations as systems for the study of calcium-triggered exocytosis. J. Physiol. 1999, 520, 15–21. [Google Scholar] [CrossRef]
- Sodergren, E.; Weinstock, G.M.; Davidson, E.H.; Cameron, R.A.; Gibbs, R.A.; Angerer, R.C.; Angerer, L.M.; Arnone, M.I.; Burgess, D.R.; Burke, R.D.; et al. The genome of the sea urchin Strongylocentrotus purpuratus. Science 2006, 314, 941–952. [Google Scholar] [CrossRef]
- Tahara, M.; Coorssen, J.R.; Timmers, K.; Blank, P.S.; Whalley, T.; Scheller, R.; Zimmerberg, J. Calcium Can Disrupt the SNARE Protein Complex on Sea Urchin Egg Secretory Vesicles without Irreversibly Blocking Fusion. J. Biol. Chem. 1998, 273, 33667–33673. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Blank, P.S.; Tahara, M.; Zimmerberg, J. Biochemical and Functional Studies of Cortical Vesicle Fusion: The SNARE Complex and Ca2+ Sensitivity. J. Cell Biol. 1998, 143, 1845–1857. [Google Scholar] [CrossRef]
- Szule, J.A.; Jarvis, S.E.; Hibbert, J.E.; Spafford, J.D.; Braun, J.E.A.; Zamponi, G.W.; Wessel, G.M.; Coorssen, J.R. Calcium-triggered Membrane Fusion Proceeds Independently of Specific Presynaptic Proteins. J. Biol. Chem. 2003, 278, 24251–24254. [Google Scholar] [CrossRef] [PubMed]
- Sean, C.; David, L.; Gary, W. Members of the SNARE hypothesis are associated with cortical granule exocytosis in the sea urchin egg. Mol. Reprod. Dev. 1997, 48, 106–118. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Blank, P.S.; Albertorio, F.; Bezrukov, L.; Kolosova, I.; Chen, X.; Backlund, P.S.; Zimmerberg, J. Regulated secretion: SNARE density, vesicle fusion and calcium dependence. J. Cell Sci. 2003, 116, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Avery, J.; Hodel, A.; Whitaker, M. In vitro exocytosis in sea urchin eggs requires a synaptobrevin-related protein. J. Cell Sci. 1997, 110, 1555–1561. [Google Scholar] [PubMed]
- Rogasevskaia, T.; Coorssen, J.R. Sphingomyelin-enriched microdomains define the efficiency of native Ca2+-triggered membrane fusion. J. Cell Sci. 2006, 119, 2688–2694. [Google Scholar] [CrossRef] [PubMed]
- Rogasevskaia, T.P.; Churchward, M.A.; Coorssen, J.R. Anionic lipids in Ca(2+)-triggered fusion. Cell Calcium 2012, 52, 259–269. [Google Scholar] [CrossRef]
- Rogasevskaia, T.P.; Coorssen, J.R. The role of phospholipase D in regulated exocytosis. J. Biol. Chem. 2015, 290, 28683–28696. [Google Scholar] [CrossRef]
- Rogasevskaia, T.P.; Coorssen, J.R. A new approach to the molecular analysis of docking, priming and regulated membrane fusion. J. Chem. Biol. 2011, 4, 117–136. [Google Scholar] [CrossRef]
- Churchward, M.A.; Rogasevskaia, T.; Höfgen, J.; Bau, J.; Coorssen, J.R. Cholesterol facilitates the native mechanism of Ca2+-triggered membrane fusion. J. Cell Sci. 2005, 118, 4833–4848. [Google Scholar] [CrossRef]
- Churchward, M.A.; Rogasevskaia, T.; Brandman, D.M.; Khosravani, H.; Nava, P.; Atkinson, J.K.; Coorssen, J.R. Specific Lipids Supply Critical Negative Spontaneous Curvature—An Essential Component of Native Ca2+-Triggered Membrane Fusion. Biophys. J. 2008, 94, 3976–3986. [Google Scholar] [CrossRef]
- Abbineni, P.; Coorssen, J. Application of High-Throughput Assays to Examine Phospho-Modulation of the Late Steps of Regulated Exocytosis. High.-Throughput 2017, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Abbineni, P.S.; Coorssen, J.R. Sphingolipids modulate docking, Ca2+ sensitivity and membrane fusion of native cortical vesicles. Int. J. Biochem. Cell Biol. 2018, 104, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Zimmerberg, J.; Blank, P.S.; Kolosova, I.; Cho, M.-S.; Tahara, M.; Coorssen, J.R. A stage-specific preparation to study the Ca2+-triggered fusion steps of exocytosis: Rationale and perspectives. Biochimie 2000, 82, 303–314. [Google Scholar] [CrossRef]
- Churchward, M.A.; Brandman, D.M.; Rogasevskaia, T.; Coorssen, J.R. Copper (II) sulfate charring for high sensitivity on-plate fluorescent detection of lipids and sterols: Quantitative analyses of the composition of functional secretory vesicles. J. Chem. Biol. 2008, 1, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Butt, R.H.; Coorssen, J.R. Postfractionation for Enhanced Proteomic Analyses: Routine Electrophoretic Methods Increase the Resolution of Standard 2D-PAGE. J. Proteome Res. 2005, 4, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.G. Building Phylogenetic Trees from Molecular Data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Blank, P.S.; Albertorio, F.; Bezrukov, L.; Kolosova, I.; Backlund, P.S., Jr.; Zimmerberg, J. Quantitative femto- to attomole immunodetection of regulated secretory vesicle proteins critical to exocytosis. Anal. Biochem. 2002, 307, 54–62. [Google Scholar] [CrossRef]
- Gauci, V.J.; Padula, M.P.; Coorssen, J.R. Coomassie blue staining for high sensitivity gel-based proteomics. J. Proteom. 2013, 90, 96–106. [Google Scholar] [CrossRef]
- Noaman, N.; Abbineni, P.S.; Withers, M.; Coorssen, J.R. Coomassie staining provides routine (sub)femtomole in-gel detection of intact proteoforms: Expanding opportunities for genuine Top-down Proteomics. Electrophoresis 2017, 38, 3086–3099. [Google Scholar] [CrossRef]
- Balsinde, J.; Bianco, I.D.; Ackermann, E.J.; Conde-Frieboes, K.; Dennis, E.A. Inhibition of calcium-independent phospholipase A2 prevents arachidonic acid incorporation and phospholipid remodeling in P388D1 macrophages. Proc. Natl. Acad. Sci. USA 1995, 92, 8527–8531. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, C.M.; Mancuso, D.J.; Yan, W.; Sims, H.F.; Gibson, B.; Gross, R.W. Identification, Cloning, Expression and Purification of Three Novel Human Calcium-independent Phospholipase A2 Family Members Possessing Triacylglycerol Lipase and Acylglycerol Transacylase Activities. J. Biol. Chem. 2004, 279, 48968–48975. [Google Scholar] [CrossRef] [PubMed]
- Farooqui, A.A.; Litsky, M.L.; Farooqui, T.; Horrocks, L.A. Inhibitors of intracellular phospholipase A2 activity: Their neurochemical effects and therapeutical importance for neurological disorders. Brain Res. Bull. 1999, 49, 139–153. [Google Scholar] [CrossRef]
- Hsu, Y.-H.; Bucher, D.; Cao, J.; Li, S.; Yang, S.-W.; Kokotos, G.; Woods, V.L.; McCammon, J.A.; Dennis, E.A. Fluoroketone Inhibition of Ca2+-Independent Phospholipase A2 through Binding Pocket Association Defined by Hydrogen/Deuterium Exchange and Molecular Dynamics. J. Am. Chem. Soc. 2013, 135, 1330–1337. [Google Scholar] [CrossRef]
- Tibes, U. Phospholipase A2 inhibitors in development. Expert Opin. Investig. Drugs 1997, 6, 279–298. [Google Scholar] [CrossRef]
- Baker, P.F.; Whitaker, M.J. Influence of ATP and calcium on the cortical reaction in sea urchin eggs. Nature 1978, 276, 513. [Google Scholar] [CrossRef] [PubMed]
- Vacquier, V.D. The isolation of intact cortical granules from sea urchin eggs: Calcium lons trigger granule discharge. Dev. Biol. 1975, 43, 62–74. [Google Scholar] [CrossRef]
- Balsinde, J.; Dennis, E.A. Bromoenol lactone inhibits magnesium-dependent phosphatidate phosphohydrolase and blocks triacylglycerol biosynthesis in mouse P388D1 macrophages. J. Biol. Chem. 1996, 271, 31937–31941. [Google Scholar] [CrossRef]
- Ramanadham, S.; Wolf, M.J.; Jett, P.A.; Gross, R.W.; Turk, J. Characterization of an ATP-stimulatable Ca(2+)-independent phospholipase A2 from clonal insulin-secreting HIT cells and rat pancreatic islets: A possible molecular component of the beta-cell fuel sensor. Biochemistry 1994, 33, 7442–7452. [Google Scholar] [CrossRef] [PubMed]
- Cameron, R.A. Comparing the Human and Sea Urchin Genomes. In eLS; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar] [CrossRef]
- Bradham, C.A.; Foltz, K.R.; Beane, W.S.; Arnone, M.I.; Rizzo, F.; Coffman, J.A.; Mushegian, A.; Goel, M.; Morales, J.; Geneviere, A.-M.; et al. The sea urchin kinome: A first look. Dev. Biol. 2006, 300, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Ramanadham, S.; Hsu, F.F.; Zhang, S.; Jin, C.; Bohrer, A.; Song, H.; Bao, S.; Ma, Z.; Turk, J. Apoptosis of insulin-secreting cells induced by endoplasmic reticulum stress is amplified by overexpression of group VIA calcium-independent phospholipase A2 (iPLA2 beta) and suppressed by inhibition of iPLA2 beta. Biochemistry 2004, 43, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Bao, S.; Lei, X.; Jin, C.; Zhang, S.; Turk, J.; Ramanadham, S. Evidence for proteolytic processing and stimulated organelle redistribution of iPLA2β. Biochim. Biophys. Acta (Bba) Mol. Cell Biol. Lipids 2010, 1801, 547–558. [Google Scholar] [CrossRef]
- Larsson, P.K.A.; Claesson, H.-E.; Kennedy, B.P. Multiple Splice Variants of the Human Calcium-independent Phospholipase A2 and Their Effect on Enzyme Activity. J. Biol. Chem. 1998, 273, 207–214. [Google Scholar] [CrossRef]
- Song, H.; Ramanadham, S.; Bao, S.; Hsu, F.-F.; Turk, J. A Bromoenol Lactone Suicide Substrate Inactivates Group VIA Phospholipase A(2) by Generating a Diffiusible Bromomethyl Keto Acid That Alkylates Cysteine Thiols. Biochemistry 2006, 45, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Darrow, A.L.; Olson, M.W.; Xin, H.; Burke, S.L.; Smith, C.; Schalk-Hihi, C.; Williams, R.; Bayoumy, S.S.; Deckman, I.C.; Todd, M.J.; et al. A novel fluorogenic substrate for the measurement of endothelial lipase activity. J. Lipid Res. 2011, 52, 374–382. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, K.P.; Rhee, H.J.; Das, S.; Rafter, J.D.; Oh, Y.S.; Cho, W. Internalized group V secretory phospholipase A2 acts on the perinuclear membranes. J. Biol. Chem. 2002, 277, 9358–9365. [Google Scholar] [CrossRef] [PubMed]
- Coorssen, J.R. Phospholipase activation and secretion: Evidence that PLA2, PLC and PLD are not essential to exocytosis. Am. J. Physiol. Cell Physiol. 1996, 270, C1153–C1163. [Google Scholar] [CrossRef] [PubMed]
- Balsinde, J.; Winstead, M.V.; Dennis, E.A. Phospholipase A2 regulation of arachidonic acid mobilization. FEBS Lett. 2002, 531, 2–6. [Google Scholar] [CrossRef]
- Peterson, M.E.; Chen, F.; Saven, J.G.; Roos, D.S.; Babbitt, P.C.; Sali, A. Evolutionary constraints on structural similarity in orthologs and paralogs. Protein Sci. A Publ. Protein Soc. 2009, 18, 1306–1315. [Google Scholar] [CrossRef]
- Hazen, S.L.; Stuppy, R.J.; Gross, R.W. Purification and characterization of canine myocardial cytosolic phospholipase A2. A calcium-independent phospholipase with absolute f1-2 regiospecificity for diradyl glycerophospholipids. J. Biol. Chem. 1990, 265, 10622–10630. [Google Scholar] [PubMed]
- Rosenthal, M.D.; Gordon, M.N.; Buescher, E.S.; Slusser, J.H.; Harris, L.K.; Franson, R.C. Human Neutrophils Store Type II 14-kDa Phospholipase A2 in Granules and Secrete Active Enzyme in Response to Soluble Stimuli. Biochem. Biophys. Res. Commun. 1995, 208, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Ishizaki, J.; Yokota, Y.; Higashino, K.; Ono, T.; Ikeda, M.; Fujii, N.; Kawamoto, K.; Hanasaki, K. Structures, enzymatic properties and expression of novel human and mouse secretory phospholipase A(2)s. J. Biol. Chem. 2000, 275, 5785–5793. [Google Scholar] [CrossRef]
- Zupan, L.A.; Steffens, D.L.; Berry, C.A.; Landt, M.; Gross, R.W. Cloning and expression of a human 14-3-3 protein mediating phospholipolysis. Identification of an arachidonoyl-enzyme intermediate during catalysis. J. Biol. Chem. 1992, 267, 8707–8710. [Google Scholar] [PubMed]
- Wilkins, M.R.; Lindskog, I.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Hochstrasser, D.F.; Appel, R.D. Detailed peptide characterization using PEPTIDEMASS—A World-Wide-Web-accessible tool. Electrophoresis 1997, 18, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Szule, J.A.; Fuller, N.L.; Rand, R.P. The effects of acyl chain length and saturation of diacylglycerols and phosphatidylcholines on membrane monolayer curvature. Biophys. J. 2002, 83, 977–984. [Google Scholar] [CrossRef]
- Furber, K.L.; Churchward, M.A.; Rogasevskaia, T.P.; Coorssen, J.R. Identifying Critical Components of Native Ca2+-triggered Membrane Fusion. Ann. N. Y. Acad. Sci. 2009, 1152, 121–134. [Google Scholar] [CrossRef]
- Furber, K.L.; Dean, K.T.; Coorssen, J.R. Dissecting the mechanism of Ca2+ triggered membrane fusion: Probing protein function using thiol reactivity. Clin. Exp. Pharmacol. Physiol. 2010, 37, 208–217. [Google Scholar] [CrossRef]
- Furber, K.L.; Brandman, D.M.; Coorssen, J.R. Enhancement of the Ca(2+)-triggering steps of native membrane fusion via thiol-reactivity. J. Chem. Biol. 2009, 2, 27–37. [Google Scholar] [CrossRef]
- Spessard, G.O. ACD Labs/LogP dB 3.5 and ChemSketch 3.5. J. Chem. Inf. Comput. Sci. 1998, 38, 1250–1253. [Google Scholar] [CrossRef]
- Chernomordik, L.; Kozlov, M.M.; Zimmerberg, J. Lipids in biological membrane fusion. J. Membarin Biol. 1995, 146, 1–14. [Google Scholar] [CrossRef]
- Chernomordik, L.V.; Kozlov, M.M. Mechanics of membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 675–683. [Google Scholar] [CrossRef]
- Kozlov, M.M.; Leikin, S.L.; Chernomordik, L.V.; Markin, V.S.; Chizmadzhev, Y.A. Stalk mechanism of vesicle fusion. Eur. Biophys. J. 1989, 17, 121–129. [Google Scholar] [CrossRef]
- Kozlovsky, Y.; Kozlov, M.M. Stalk Model of Membrane Fusion: Solution of Energy Crisis. Biophys. J. 2002, 82, 882–895. [Google Scholar] [CrossRef]
- Fuller, N.; Rand, R.P. The influence of lysolipids on the spontaneous curvature and bending elasticity of phospholipid membranes. Biophys. J. 2001, 81, 243–254. [Google Scholar] [CrossRef]
- Gillot, I.; Ciapa, B.; Payan, P.; Sardet, C. The calcium content of cortical granules and the loss of calcium from sea urchin eggs at fertilization. Dev. Biol. 1991, 146, 396–405. [Google Scholar] [CrossRef]
- Mahapatra, N.R.; Mahata, M.; Hazra, P.P.; McDonough, P.M.; O’Connor, D.T.; Mahata, S.K. A Dynamic Pool of Calcium in Catecholamine Storage Vesicles: Exploration in living cells by a novel vesicle-targeted chromogranin a-aequorin chimeric photoprotein. J. Biol. Chem. 2004, 279, 51107–51121. [Google Scholar] [CrossRef] [PubMed]
- Leitner, J.W.; Sussman, K.E.; Vatter, A.E.; Schneider, F.H. Adenine nucleotides in the secretory granule fraction of rat islets. Endocrinology 1975, 96, 662–677. [Google Scholar] [CrossRef] [PubMed]
- Brash, A.R. Arachidonic acid as a bioactive molecule. J. Clin. Investig. 2001, 107, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A. Fatty acid transport: Difficult or easy? J. Lipid Res. 1998, 39, 467–481. [Google Scholar]
- Alder-Baerens, N.; Lisman, Q.; Luong, L.; Pomorski, T.; Holthuis, J.C.M. Loss of P4 ATPases Drs2p and Dnf3p Disrupts Aminophospholipid Transport and Asymmetry in Yeast Post-Golgi Secretory Vesicles. Mol. Biol. Cell 2006, 17, 1632–1642. [Google Scholar] [CrossRef] [PubMed]
- Galli, C.; Risé, P.; Marangoni, F. Fate of exogenous arachidonic acid in THP-1 cells: Incorporation in cell lipids and conversion to other N-6 fatty acids. ProstaglandinsLeukot. Essent. Fat. Acids 1995, 52, 103–106. [Google Scholar] [CrossRef]
- Ibarguren, M.; López, D.J.; Escribá, P.V. The effect of natural and synthetic fatty acids on membrane structure, microdomain organization, cellular functions and human health. Biochim. Biophys. Acta (Bba) Biomembr. 2014, 1838, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Epand, R.F.; Ahmed, N.; Chen, R. Promotion of hexagonal phase formation and lipid mixing by fatty acids with varying degrees of unsaturation. Chem. Phys. Lipids 1991, 57, 75–80. [Google Scholar] [CrossRef]
- Darios, F.; Davletov, B. Omega-3 and omega-6 fatty acids stimulate cell membrane expansion by acting on syntaxin 3. Nature 2006, 440, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Dulubova, I.; Sugita, S.; Hill, S.; Hosaka, M.; Fernandez, I.; Sudhof, T.C.; Rizo, J. A conformational switch in syntaxin during exocytosis: Role of munc18. EMBO J. 1999, 18, 4372–4382. [Google Scholar] [CrossRef] [PubMed]
- Chernomordik, L. Non-bilayer lipids and biological fusion intermediates. Chem. Phys. Lipids 1996, 81, 203–213. [Google Scholar] [CrossRef]
Figure 1.
Detection and quantification of (A) endogenous CSC FFA, n = 3 separate experiments; (B) NBD-FFA and -PA in intact CSC after 15 min labelling with NBD-PC followed by 20 min inhibitor treatments, n = 3 separate experiments; (C) total CSC neutral lipids visualized on the same TLC plate as shown in B, after charring with CuSO4. This also confirmed consistent lipid loading in each lane. Representative chromatograms showing indicated changes in lipids. (p-value, * < 0.05, ** < 0.005, **** < 0.0001 indicates relative difference to the control). Note: A section of chromatogram is shown in A and lanes in all chromatograms are grouped together from the same HPTLC plate following removal of lanes not associated with this study. A standard neutral lipid protocol was used to resolve the lipids [56].
Figure 1.
Detection and quantification of (A) endogenous CSC FFA, n = 3 separate experiments; (B) NBD-FFA and -PA in intact CSC after 15 min labelling with NBD-PC followed by 20 min inhibitor treatments, n = 3 separate experiments; (C) total CSC neutral lipids visualized on the same TLC plate as shown in B, after charring with CuSO4. This also confirmed consistent lipid loading in each lane. Representative chromatograms showing indicated changes in lipids. (p-value, * < 0.05, ** < 0.005, **** < 0.0001 indicates relative difference to the control). Note: A section of chromatogram is shown in A and lanes in all chromatograms are grouped together from the same HPTLC plate following removal of lanes not associated with this study. A standard neutral lipid protocol was used to resolve the lipids [56].

Figure 2.
Effects of LY311727 (selective for sPLA2) and BEL and FKGK-11 (both selective for iPLA2) on (A) CSC Ca2+ activity curves (i.e., exocytosis in vitro); (B) fusion extent and; (C) Ca2+ sensitivity (EC50); n = 3–14. (p-value, * < 0.05, *** < 0.0005, **** < 0.0001 indicates difference relative to the control).
Figure 2.
Effects of LY311727 (selective for sPLA2) and BEL and FKGK-11 (both selective for iPLA2) on (A) CSC Ca2+ activity curves (i.e., exocytosis in vitro); (B) fusion extent and; (C) Ca2+ sensitivity (EC50); n = 3–14. (p-value, * < 0.05, *** < 0.0005, **** < 0.0001 indicates difference relative to the control).

Figure 3.
Sequence alignment of predicted urchin (A) sPLA2 and (B) 80–85 kDa iPLA2. The first row in each panel is the amino acid sequence of urchin PLA2 isozymes and the subsequent two rows are of murine and human PLA2 isozymes, respectively. Conserved amino acids are indicated with an asterisk and site # represents the amino acid residue number in the primary urchin PLA2 sequence (also considering gaps). The red arrow indicates conserved Histidine in the Ca2+ binding loop (Black box), the site at which LY311727 binds. Black arrows show conserved Histidine and Aspartic acid which form the catalytic site His-Asp dyad. Blue and red boxes indicate the caspase cleavage site and the active site serine in the GXSXG consensus sequence, respectively. The circle indicates the nucleotide binding site. Dissimilar residues with the same background colour indicate conservative substitution.
Figure 3.
Sequence alignment of predicted urchin (A) sPLA2 and (B) 80–85 kDa iPLA2. The first row in each panel is the amino acid sequence of urchin PLA2 isozymes and the subsequent two rows are of murine and human PLA2 isozymes, respectively. Conserved amino acids are indicated with an asterisk and site # represents the amino acid residue number in the primary urchin PLA2 sequence (also considering gaps). The red arrow indicates conserved Histidine in the Ca2+ binding loop (Black box), the site at which LY311727 binds. Black arrows show conserved Histidine and Aspartic acid which form the catalytic site His-Asp dyad. Blue and red boxes indicate the caspase cleavage site and the active site serine in the GXSXG consensus sequence, respectively. The circle indicates the nucleotide binding site. Dissimilar residues with the same background colour indicate conservative substitution.

Figure 4.
Immuno-reactive spots detected by (A) sPLA2 antibody in the CV luminal fraction; (B) iPLA2 antibody in the CSC membrane fraction; and (C) iPLA2 antibody in the CV membrane fraction. cCBB stained 2D SDS-PAGE gels (left) and parallel western blots (right).
Figure 4.
Immuno-reactive spots detected by (A) sPLA2 antibody in the CV luminal fraction; (B) iPLA2 antibody in the CSC membrane fraction; and (C) iPLA2 antibody in the CV membrane fraction. cCBB stained 2D SDS-PAGE gels (left) and parallel western blots (right).
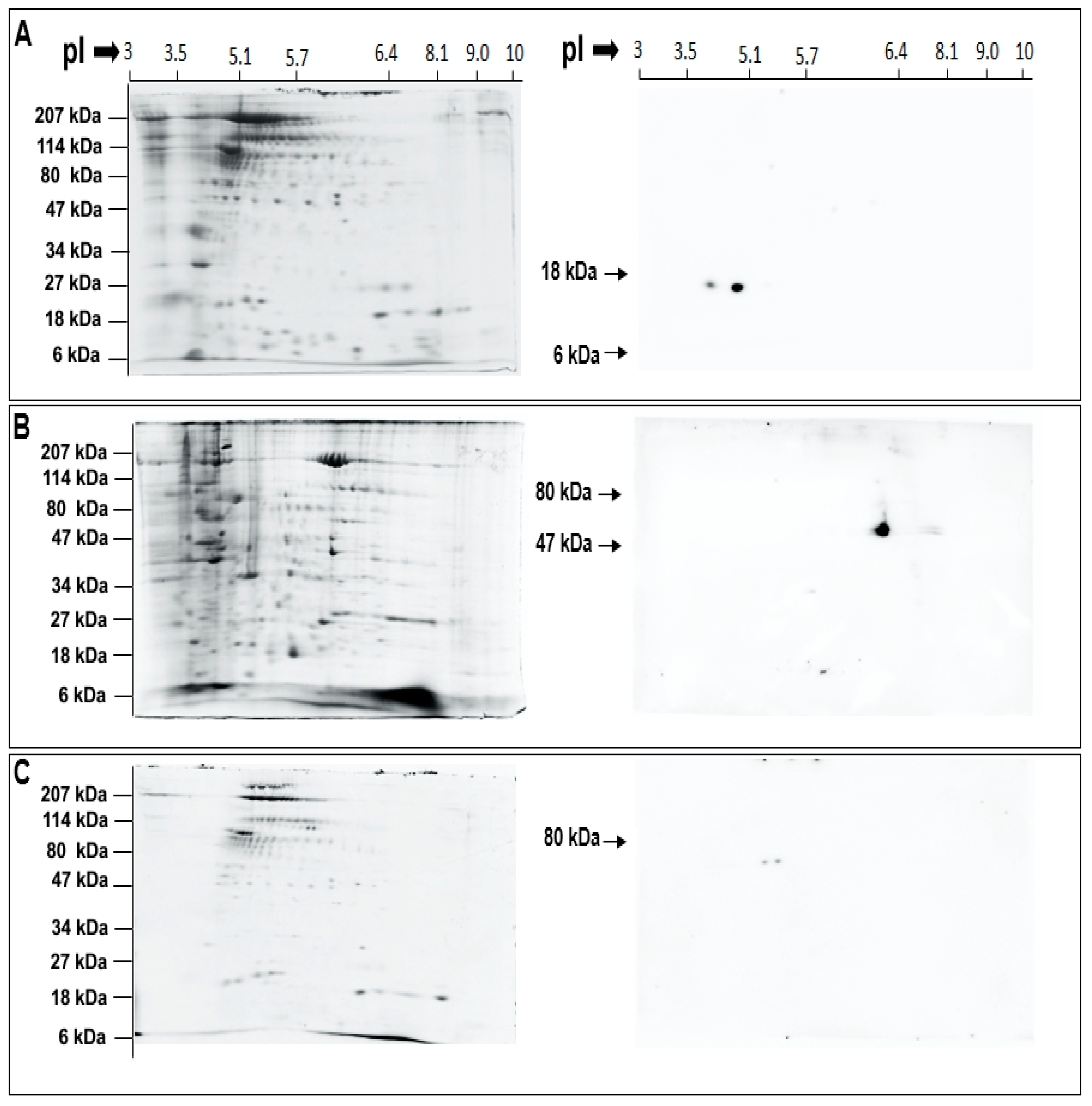
Figure 5.
(A) Time-dependent decrease in NBD-PE and no change in NBD-PC, when incubated with isolated CV luminal proteins, n = 3; (B) Detection of NBD-FFA generated from NBD-PE, when incubated with CV luminal proteins; n = 2–3. After incubations, activity was stopped by adding 0.2 µL HCl, total lipids isolated, resulting organic and aqueous phases retained, dried and resolved. (C) Bar graph summarizing the changes in NBD-PC and -PE, indicating the presence of CV luminal PLA2 with high catalytic activity for PE. (D) Effect of LY311727, increased [Ca2+]free and boiling on luminal PLA2 activity assessed by incubating isolated CV luminal proteins with NBD-PE; n = 3. (p-value, * < 0.05 indicates difference relative to the control). The chromatograms in A were resolved using a standard phospholipid protocol and in (B,D) using an established protocol for neutral lipids [56]. Note: Left panels in (A,B) show NBD fluorescence and the right panels show same section of the chromatogram after charring with CuSO4 [56]. The lanes shown in (D) are grouped together from the same chromatogram following removal of lanes not associated with this study.
Figure 5.
(A) Time-dependent decrease in NBD-PE and no change in NBD-PC, when incubated with isolated CV luminal proteins, n = 3; (B) Detection of NBD-FFA generated from NBD-PE, when incubated with CV luminal proteins; n = 2–3. After incubations, activity was stopped by adding 0.2 µL HCl, total lipids isolated, resulting organic and aqueous phases retained, dried and resolved. (C) Bar graph summarizing the changes in NBD-PC and -PE, indicating the presence of CV luminal PLA2 with high catalytic activity for PE. (D) Effect of LY311727, increased [Ca2+]free and boiling on luminal PLA2 activity assessed by incubating isolated CV luminal proteins with NBD-PE; n = 3. (p-value, * < 0.05 indicates difference relative to the control). The chromatograms in A were resolved using a standard phospholipid protocol and in (B,D) using an established protocol for neutral lipids [56]. Note: Left panels in (A,B) show NBD fluorescence and the right panels show same section of the chromatogram after charring with CuSO4 [56]. The lanes shown in (D) are grouped together from the same chromatogram following removal of lanes not associated with this study.
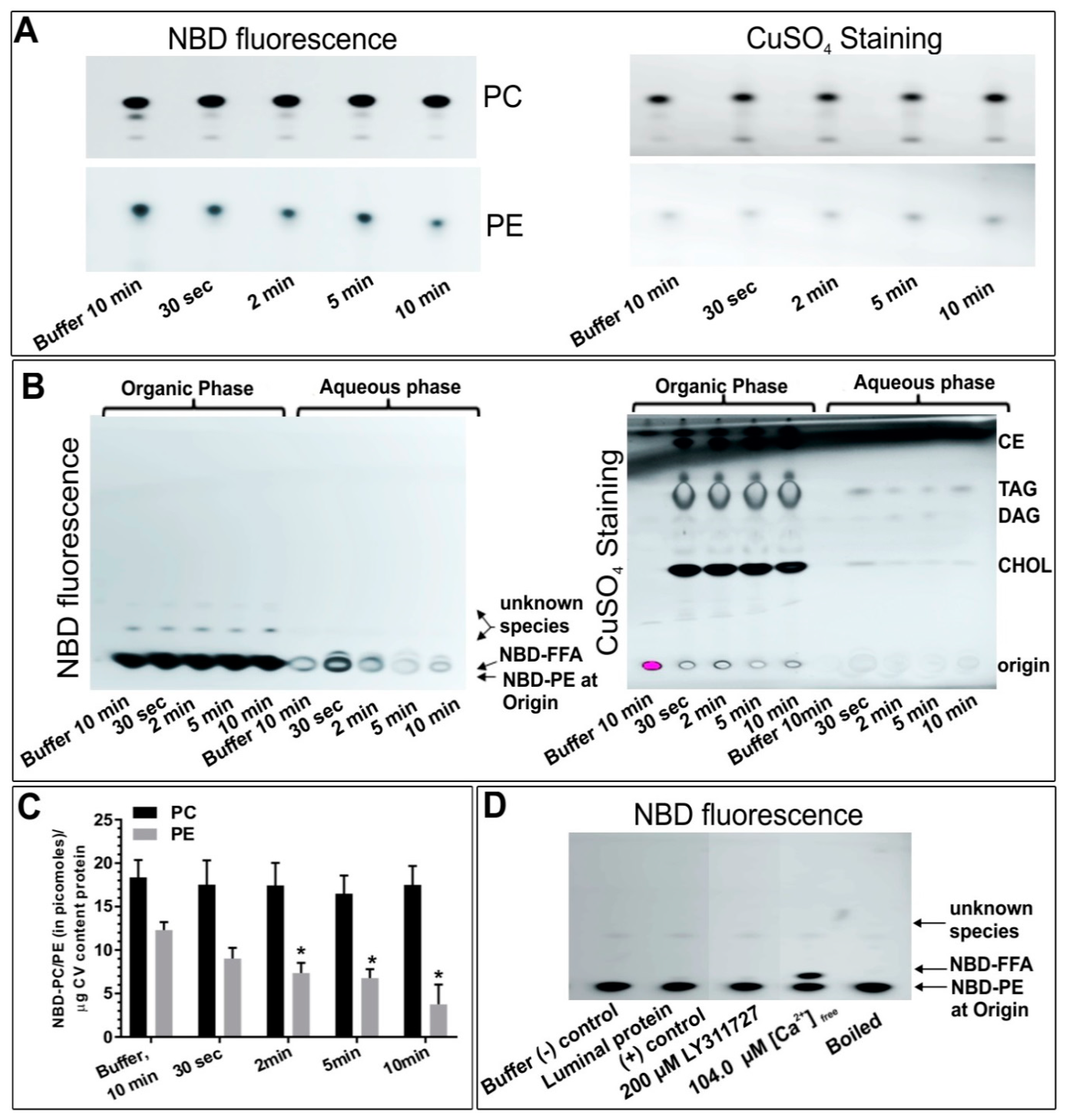
Figure 6.
(A) Time-dependent changes in NBD-PC and -PE, when incubated with CV membrane fragments (n = 2–3); (B) Western blot showing an apparent complete loss of iPLA2 in trypsin treated CV (n = 3). A total of 5µg CV membrane protein per lane was resolved; (C) Time-dependent changes in the CV surface PLA2 activity in the intact CV assessed using PLA2-selective substrate PED6 [79,80] (n = 3). The loss of the fluorescence signal is consistent with the loss of PLA2 from the CV surface following ablation with trypsin. Functional assays showing the effects of trypsin treatment on CV-CV Ca2+ activity curves [45]; (D) Standard fusion assay (n = 2); (E) CV settle fusion assay (n = 3). (p-value, * < 0.05, ** < 0.005 indicates difference relative to the control). Note: The lanes in (B) are grouped together from the same western blot following removal of intervening lanes.
Figure 6.
(A) Time-dependent changes in NBD-PC and -PE, when incubated with CV membrane fragments (n = 2–3); (B) Western blot showing an apparent complete loss of iPLA2 in trypsin treated CV (n = 3). A total of 5µg CV membrane protein per lane was resolved; (C) Time-dependent changes in the CV surface PLA2 activity in the intact CV assessed using PLA2-selective substrate PED6 [79,80] (n = 3). The loss of the fluorescence signal is consistent with the loss of PLA2 from the CV surface following ablation with trypsin. Functional assays showing the effects of trypsin treatment on CV-CV Ca2+ activity curves [45]; (D) Standard fusion assay (n = 2); (E) CV settle fusion assay (n = 3). (p-value, * < 0.05, ** < 0.005 indicates difference relative to the control). Note: The lanes in (B) are grouped together from the same western blot following removal of intervening lanes.

Figure 7.
Effects of LY311727 (selective for sPLA2) and BEL and FKGK-11 (both selective for iPLA2) on CV-CV Ca2+ activity curves, fusion extent and Ca2+ sensitivity (EC50); (A) standard fusion assay, n = 3–19; (B) settle fusion assay, n = 3–19. (p-value, * < 0.05, ** < 0.005, *** < 0.0005, **** < 0.0001 indicates difference relative to the control.
Figure 7.
Effects of LY311727 (selective for sPLA2) and BEL and FKGK-11 (both selective for iPLA2) on CV-CV Ca2+ activity curves, fusion extent and Ca2+ sensitivity (EC50); (A) standard fusion assay, n = 3–19; (B) settle fusion assay, n = 3–19. (p-value, * < 0.05, ** < 0.005, *** < 0.0005, **** < 0.0001 indicates difference relative to the control.

Figure 8.
(A) Changes in the endogenous FFA in CV treated with indicated doses of LY311727, BEL and FKGK-11; n = 4–12; (B) Section of the chromatograms showing the changes in endogenous FFA. (p-value, * < 0.05, ** < 0.005 indicates difference relative to the control). A standard neutral lipid protocol was used to resolve the lipids [56].
Figure 8.
(A) Changes in the endogenous FFA in CV treated with indicated doses of LY311727, BEL and FKGK-11; n = 4–12; (B) Section of the chromatograms showing the changes in endogenous FFA. (p-value, * < 0.05, ** < 0.005 indicates difference relative to the control). A standard neutral lipid protocol was used to resolve the lipids [56].
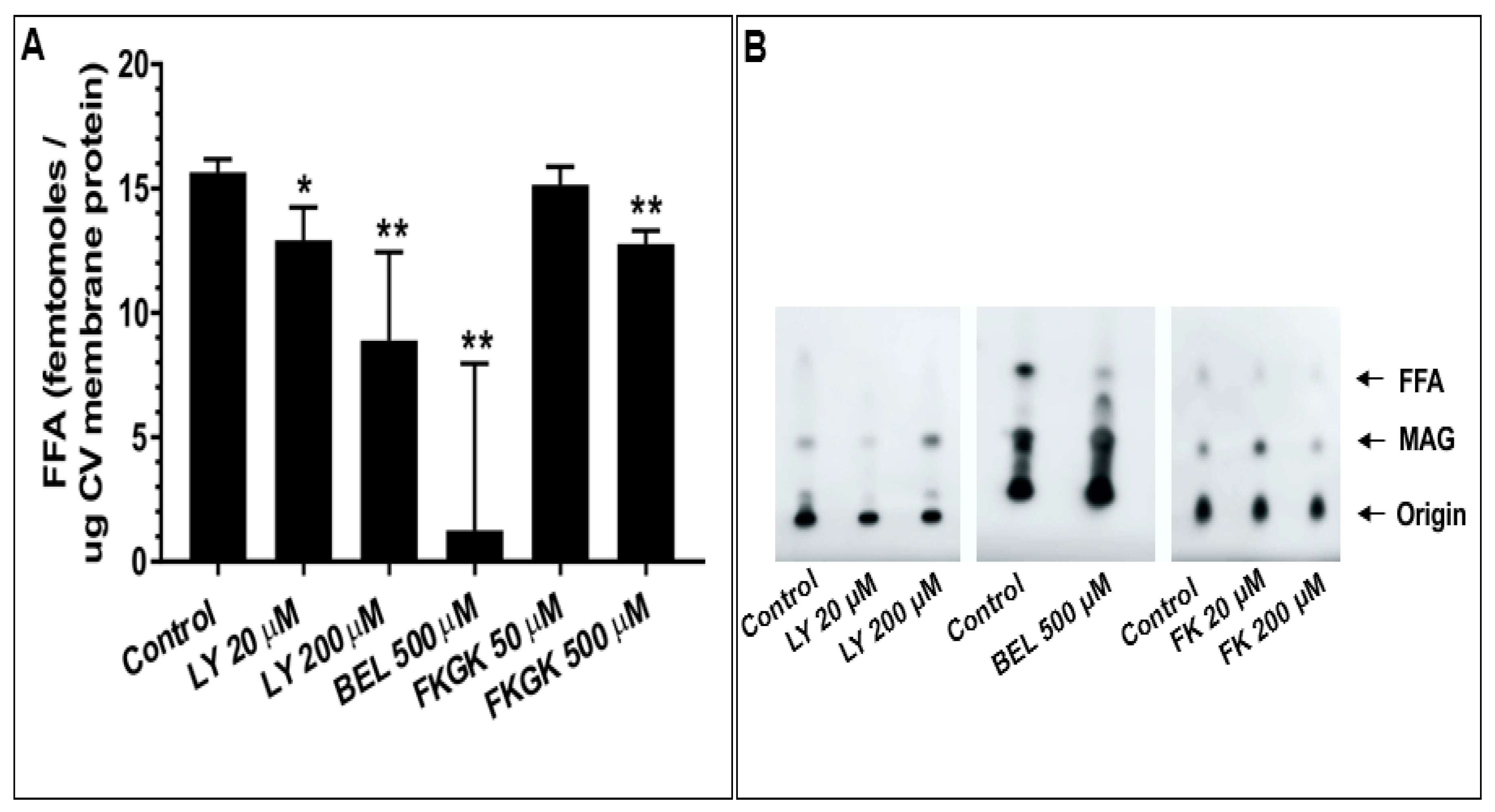
Figure 9.
Working hypothesis: (A) Non-contacting membranes (B) Vesicle docked with the plasma membrane (PM). At basal rate, the vesicle luminal sPLA2 and membrane bound iPLA2 cleave PE and PC in the inner and outer monolayer, respectively; PM bound iPLA2 also cleaves PC in the inner monolayer of the PM. This causes a local increase in LPC at the membrane contact site that is balanced by intermittent LPE production at the adjacent inner membrane via transient sPLA2 activity. This mechanism may thus act as a native fusion ‘brake’ and the released FFA (often Arachidonic acid) may support trans SNARE complex formation to ensure and maintain efficient priming/docking. (adapted from [29] with permission).
Figure 9.
Working hypothesis: (A) Non-contacting membranes (B) Vesicle docked with the plasma membrane (PM). At basal rate, the vesicle luminal sPLA2 and membrane bound iPLA2 cleave PE and PC in the inner and outer monolayer, respectively; PM bound iPLA2 also cleaves PC in the inner monolayer of the PM. This causes a local increase in LPC at the membrane contact site that is balanced by intermittent LPE production at the adjacent inner membrane via transient sPLA2 activity. This mechanism may thus act as a native fusion ‘brake’ and the released FFA (often Arachidonic acid) may support trans SNARE complex formation to ensure and maintain efficient priming/docking. (adapted from [29] with permission).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Identity and similarity of urchin PLA2 isozymes to the human and murine forms.
| Target Species | Percent Identities | Percent Similarities | Percent Identities | Percent Similarities | |
|---|---|---|---|---|---|
| Urchin Isozymes | (Full Length) | (at Histidine Catalytic Site) | |||
| sPLA2 | Human | 39 | 50 | 68 | 67 |
| Murine | 36 | 46 | 74 | 74 | |
| (Full Length) | (at Serine Catalytic Site) | ||||
| iPLA2 | Human | 36 | 52 | 100 | 100 |
| Murine | 36 | 53 | 100 | 100 | |
Table 2.
Endogenous PE and PC levels in (A) CSC and (B) CV, treated with the indicated doses of the inhibitors. ND-not done; NA-not applicable.
Table 2.
Endogenous PE and PC levels in (A) CSC and (B) CV, treated with the indicated doses of the inhibitors. ND-not done; NA-not applicable.
| A | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PE (Picomoles/µg CSC Membrane Protein) | Mean | SEM | Student t-Test | Sample Size (n) | PC (Picomoles/µg CSC Membrane Protein) | Mean | SEM | Student t-Test | Sample Size (n) | ||
| Control | 29.5 | 7.5 | 5 | Control | 47.7 | 11.0 | 6 | ||||
| LY311727 | 20 µM | ND | NA | NA | NA | LY311727 | 20 µM | 66.6 | 3.0 | 0.38 | 2 |
| LY311727 | 200 µM | ND | NA | NA | NA | LY311727 | 200 µM | 38.9 | 2.2 | 0.68 | 2 |
| BEL | 100 µM | ND | NA | NA | NA | BEL | 100 µM | ND | ND | NA | NA |
| BEL | 500 µM | 28.0 | 7.5 | 0.90 | 3 | BEL | 500 µM | 77.2 | 5.0 | 0.31 | 3 |
| FKGK-11 | 50 µM | 51.0 | 7.6 | 0.11 | 3 | FKGK-11 | 50 µM | 63.2 | 19.1 | 0.47 | 3 |
| FKGK-11 | 200 µM | 31.8 | 9.3 | 0.85 | 3 | FKGK-11 | 200 µM | 50.7 | 8.9 | 0.86 | 3 |
| B | |||||||||||
| PE (Picomoles/µg CV Membrane Protein) | Mean | SEM | Student t-Test | Sample Size (n) | PC (Picomoles/µg CV Membrane Protein) | Mean | SEM | Student t-Test | Sample Size (n) | ||
| Control | 46.0 | 5.5 | 11 | Control | 75.1 | 11.8 | 13 | ||||
| LY311727 | 20 µM | ND | NA | NA | NA | LY311727 | 20 µM | 96.4 | 26.3 | 0.5 | 3 |
| LY311727 | 200 µM | 70.5 | 5.1 | 0.05 | 3 | LY311727 | 200 µM | 114.2 | 19.9 | 0.1 | 6 |
| BEL | 100 µM | ND | NA | NA | 1 | BEL | 100 µM | ND | NA | NA | NA |
| BEL | 500 µM | 46.6 | 12.7 | 0.96 | 5 | BEL | 500 µM | 57.3 | 7.2 | 0.4 | 5 |
| FKGK-11 | 50 µM | 40.4 | 12.9 | 0.66 | 3 | FKGK-11 | 50 µM | 53.5 | 26.0 | 0.4 | 3 |
| FKGK-11 | 200 µM | 22.5 | 6.7 | 0.06 | 3 | FKGK-11 | 200 µM | 42.4 | 16.5 | 0.2 | 3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dabral, D.; Coorssen, J.R. Combined Targeted Omic and Functional Assays Identify Phospholipases A2 that Regulate Docking/Priming in Calcium-Triggered Exocytosis. Cells 2019, 8, 303. https://doi.org/10.3390/cells8040303
AMA Style
Dabral D, Coorssen JR. Combined Targeted Omic and Functional Assays Identify Phospholipases A2 that Regulate Docking/Priming in Calcium-Triggered Exocytosis. Cells. 2019; 8(4):303. https://doi.org/10.3390/cells8040303
Chicago/Turabian StyleDabral, Deepti, and Jens R Coorssen. 2019. "Combined Targeted Omic and Functional Assays Identify Phospholipases A2 that Regulate Docking/Priming in Calcium-Triggered Exocytosis" Cells 8, no. 4: 303. https://doi.org/10.3390/cells8040303
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.