An Emerin LEM-Domain Mutation Impairs Cell Response to Mechanical Stress

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.3. Self-Assembly Kinetics Followed by Thioflavin T (ThT) Fluorescence
2.4. Negative-Staining Electron Microscopy
2.5. X-Ray Crystallography
2.6. Size-Exclusion Chromatography (SEC)
2.7. Cell Culture and Reagents
2.8. Cell Plating on Substrates of Different Stiffness
2.9. Cyclic Strain
2.10. Antibodies
2.11. Immunohistochemistry and Immunofluorescence Microscopy
2.12. Protein Extraction and Immunoblotting
2.13. qPCR
2.14. Image Analysis
2.15. Statistics
3. Results
3.1. The Three Emerin Mutations Associated With Cardiac Defects Favor Emerin Self-Assembly In Vitro
3.2. Mutation ΔK37, Most Commonly Found in Patients With ACD, Causes Emerin Degradation in the Cell
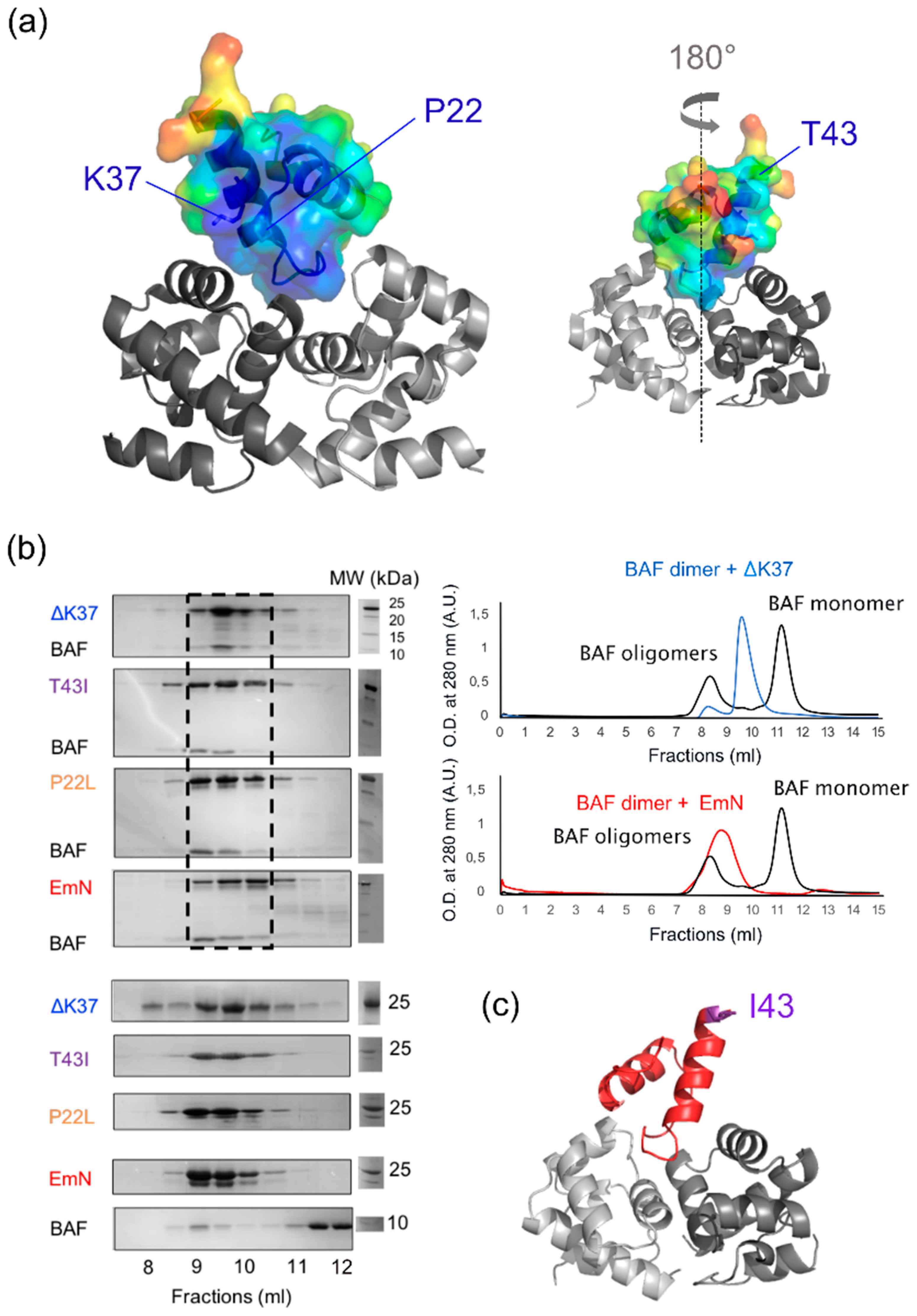
3.3. The Three Emerin Mutants Interact With BAF in Vitro
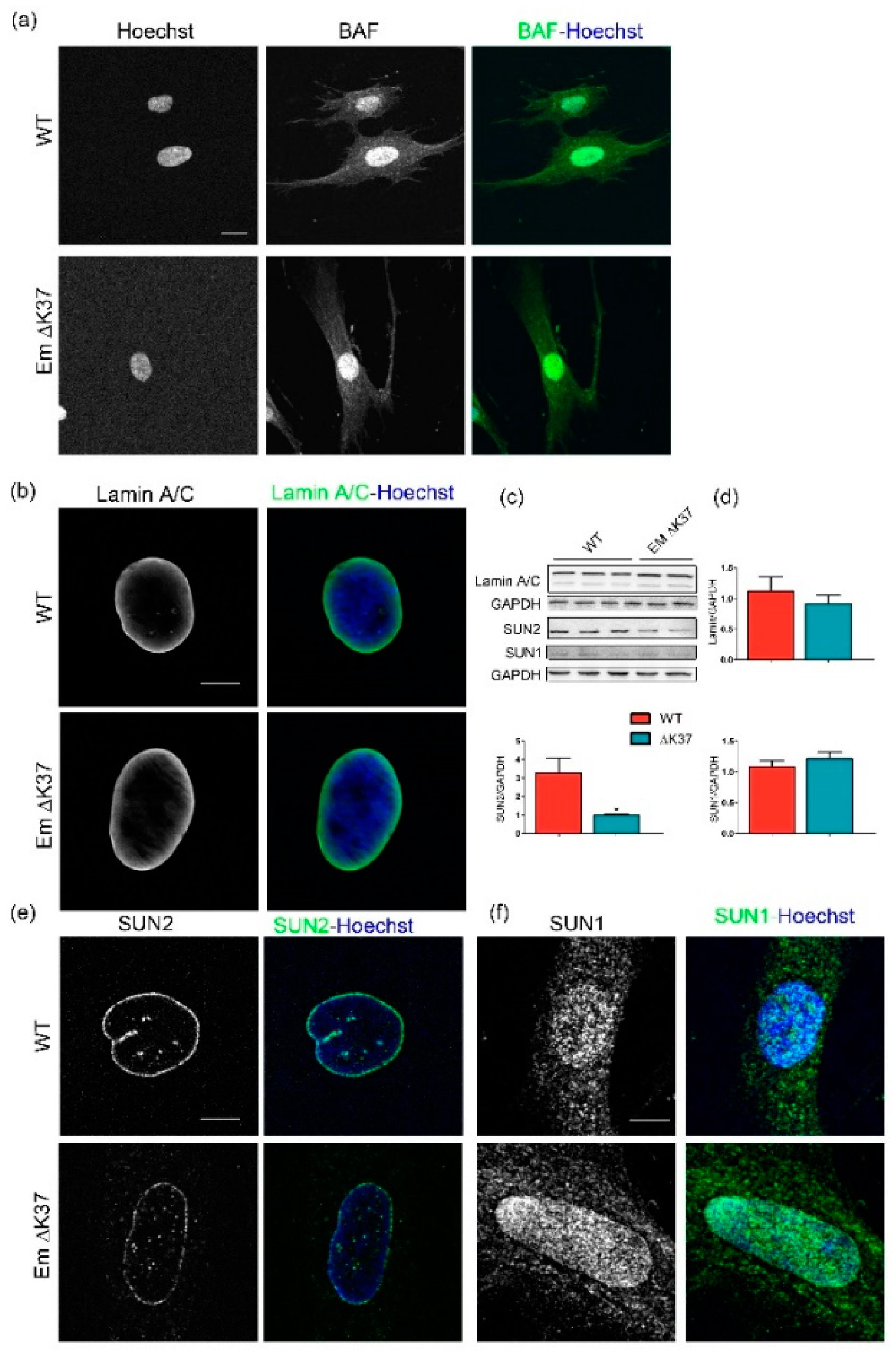
3.4. Mutation ΔK37 Does Not Impact Levels of Lamin A/C and SUN1 Nor Their Localization, But Causes a Significant Decrease in SUN2 Level
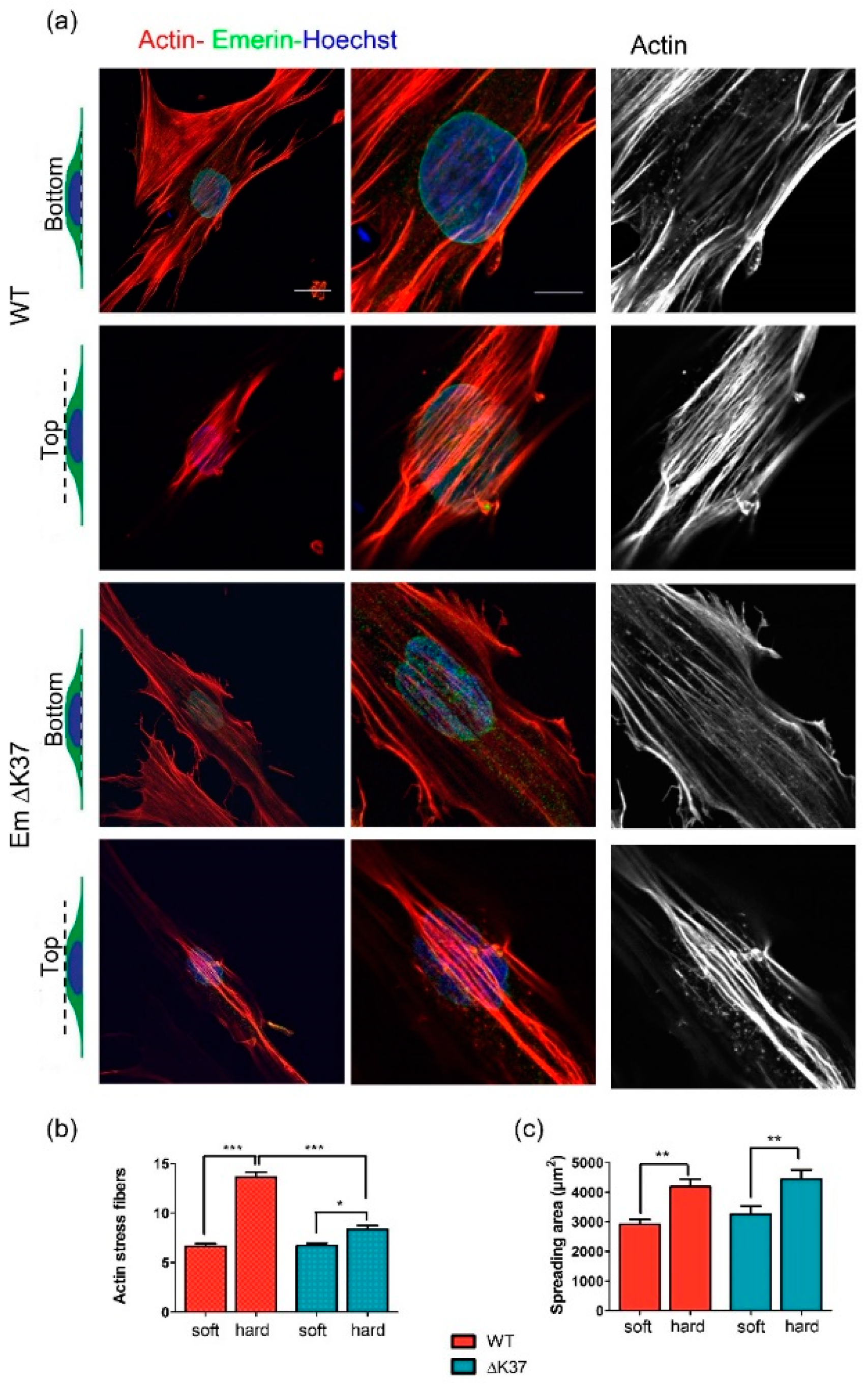
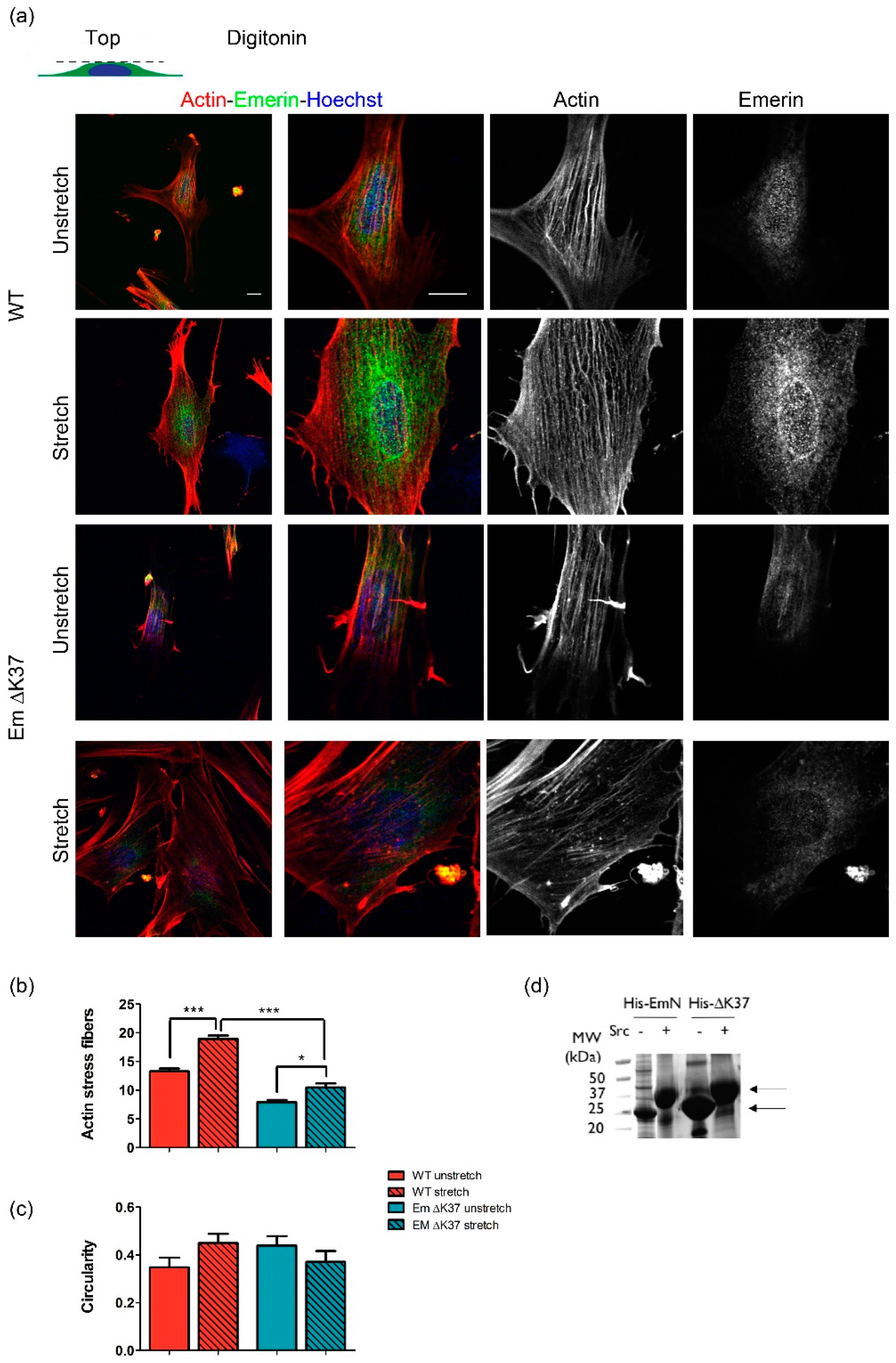
3.5. Mutation ΔK37 Impairs the Cell Response to Substrate Stiffness and Cyclic Stretch
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manilal, S.; Nguyen thi Man, C.A.; Morris, G.E. The Emery-Dreifuss Muscular Dystrophy Protein, Emerin, is a Nuclear Membrane Protein. Hum. Mol. Genet. 1996, 5, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Nagano, A.; Koga, R.; Ogawa, M.; Kurano, Y.; Kawada, J.; Okada, R.; Hayashi, Y.K.; Tsukahara, T.; Arahata, K. Emerin deficiency at the nuclear membrane in patients with Emery-Dreifuss muscular dystrophy. Nat. Genet. 1996, 12, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Bione, S.; Maestrini, E.; Rivella, S.; Mancini, M.; Regis, S.; Romeo, G.; Toniolo, D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat. Genet. 1994, 8, 323. [Google Scholar] [CrossRef] [PubMed]
- Berk, J.M.; Simon, D.N.; Jenkins-Houk, C.R.; Westerbeck, J.W.; Gronning-Wang, L.M.; Carlson, C.R.; Wilson, K.L. The molecular basis of emerin-emerin and emerin-BAF interactions. J. Cell Sci. 2014, 127, 3956–3969. [Google Scholar] [CrossRef] [PubMed]
- Wolff, N.; Gilquin, B.; Courchay, K.; Callebaut, I.; Worman, H.J.; Zinn-Justin, S. Structural analysis of emerin, an inner nuclear membrane protein mutated in X-linked Emery–Dreifuss muscular dystrophy. FEBS Lett. 2001, 501, 171–176. [Google Scholar] [CrossRef]
- Berk, J.M.; Tifft, K.E.; Wilson, K.L. The nuclear envelope LEM-domain protein emerin. Nucleus 2013, 4, 298–314. [Google Scholar] [CrossRef] [Green Version]
- Holaska, J.M.; Wilson, K.L. Multiple roles for emerin: Implications for Emery-Dreifuss muscular dystrophy. Anat. Rec. A. Discov. Mol. Cell. Evol. Biol. 2006, 288, 676–680. [Google Scholar] [CrossRef]
- UMD-EMD Database. Available online: http://www.umd.be/EMD/ (accessed on 1 June 2019).
- Bonne, G.; Leturcq, F.; Ben Yaou, R. Emery-Dreifuss Muscular Dystrophy. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Maraldi, N.M.; Mattioli, E.; Lattanzi, G.; Columbaro, M.; Capanni, C.; Camozzi, D.; Squarzoni, S.; Manzoli, F.A. Prelamin A processing and heterochromatin dynamics in laminopathies. Adv. Enzyme Regul. 2007, 47, 154–167. [Google Scholar] [CrossRef]
- Capanni, C.; Squarzoni, S.; Cenni, V.; D’Apice, M.R.; Gambineri, A.; Novelli, G.; Wehnert, M.; Pasquali, R.; Maraldi, N.M.; Lattanzi, G. Familial partial lipodystrophy, mandibuloacral dysplasia and restrictive dermopathy feature barrier-to-autointegration factor (BAF) nuclear redistribution. Cell Cycle 2012, 11, 3568–3577. [Google Scholar] [CrossRef] [Green Version]
- Iyer, A.; Koch, A.J.; Holaska, J.M. Expression Profiling of Differentiating Emerin-Null Myogenic Progenitor Identifies Molecular Pathways Implicated in Their Impaired Differentiation. Cells 2017, 6, 38. [Google Scholar] [CrossRef]
- Collins, C.M.; Ellis, J.A.; Holaska, J.M. MAPK signaling pathways and HDAC3 activity are disrupted during differentiation of emerin-null myogenic progenitor cells. Dis. Model. Mech. 2017, 10, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.J.; Holaska, J.M. Loss of Emerin Alters Myogenic Signaling and miRNA Expression in Mouse Myogenic Progenitors. PLoS ONE 2012, 7, e37262. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; Pavlidis, P.; Bonne, G.; Hayashi, Y.K.; Worman, H.J. Activation of MAPK in hearts of EMD null mice: Similarities between mouse models of X-linked and autosomal dominant Emery–Dreifuss muscular dystrophy. Hum. Mol. Genet. 2007, 16, 1884–1895. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, E.; Tilgner, K.; Barker, N.; van de Wetering, M.; Clevers, H.; Dorobek, M.; Hausmanowa-Petrusewicz, I.; Ramaekers, F.C.; Broers, J.L.; Blankesteijn, W.M.; et al. The inner nuclear membrane protein Emerin regulates β-catenin activity by restricting its accumulation in the nucleus. EMBO J. 2006, 25, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Stubenvoll, A.; Rice, M.; Wietelmann, A.; Wheeler, M.; Braun, T. Attenuation of Wnt/β-catenin activity reverses enhanced generation of cardiomyocytes and cardiac defects caused by the loss of emerin. Hum. Mol. Genet. 2015, 24, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Salpingidou, G.; Smertenko, A.; Hausmanowa-Petrucewicz, I.; Hussey, P.J.; Hutchison, C.J. A novel role for the nuclear membrane protein emerin in association of the centrosome to the outer nuclear membrane. J. Cell Biol. 2007, 178, 897–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilluy, C.; Osborne, L.D.; Van Landeghem, L.; Sharek, L.; Superfine, R.; Garcia-Mata, R.; Burridge, K. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell Biol. 2014, 16, 376–381. [Google Scholar] [CrossRef]
- Le, H.Q.; Ghatak, S.; Yeung, C.Y.; Tellkamp, F.; Günschmann, C.; Dieterich, C.; Yeroslaviz, A.; Habermann, B.; Pombo, A.; Niessen, C.M.; et al. Mechanical regulation of transcription controls Polycomb-mediated gene silencing during lineage commitment. Nat. Cell Biol. 2016, 18, 864–875. [Google Scholar] [CrossRef]
- Lee, K.K.; Haraguchi, T.; Lee, R.S.; Koujin, T.; Hiraoka, Y.; Wilson, K.L. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J. Cell Sci. 2001, 114, 4567–4573. [Google Scholar]
- Holaska, J.M.; Kowalski, A.K.; Wilson, K.L. Emerin Caps the Pointed End of Actin Filaments: Evidence for an Actin Cortical Network at the Nuclear Inner Membrane. PLoS Biol. 2004, 2, e231. [Google Scholar] [CrossRef]
- Pradhan, R.; Ranade, D.; Sengupta, K. Emerin modulates spatial organization of chromosome territories in cells on softer matrices. Nucleic Acids Res. 2018, 46, 5561–5586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Yaou, R.; Gerard, M.; Chami, K.; Sehier, A.; Belin, A.; Labombarda, F.; Richard, P.; Bonne, G.; Leturcq, F.; Chapon, F.; et al. G.P.142: A new EMD gene missense mutation in exon 1 leads to absence of emerin and is responsible for X-linked dilated cardiomyopathy with conduction defects and arrhythmias and almost elusive skeletal muscle features. Neuromuscul. Disord. 2014, 24, 843–844. [Google Scholar] [CrossRef]
- Yaou, R.B.; Toutain, A.; Arimura, T.; Demay, L.; Massart, C.; Peccate, C.; Muchir, A.; Llense, S.; Deburgrave, N.; Leturcq, F.; et al. Multitissular involvement in a family with LMNA and EMD mutations: Role of digenic mechanism? Neurology 2007, 68, 1883–1894. [Google Scholar] [CrossRef] [PubMed]
- Karst, M.L.; Herron, K.J.; Olson, T.M. X-Linked Nonsyndromic Sinus Node Dysfunction and Atrial Fibrillation Caused by Emerin Mutation. J. Cardiovasc. Electrophysiol. 2008, 19, 510–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samson, C.; Celli, F.; Hendriks, K.; Zinke, M.; Essawy, N.; Herrada, I.; Arteni, A.A.; Theillet, F.X.; Alpha-Bazin, B.; Armengaud, J.; et al. Emerin self-assembly mechanism: Role of the LEM domain. FEBS J. 2017, 284, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Huang, Y.; Suh, J.-Y.; Louis, J.M.; Ghirlando, R.; Craigie, R.; Clore, G.M. Solution NMR Structure of the Barrier-to-Autointegration Factor-Emerin Complex. J. Biol. Chem. 2007, 282, 14525–14535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, C.M.; Ronning, D.R.; Ghirlando, R.; Craigie, R.; Dyda, F. Structural basis for DNA bridging by barrier-to-autointegration factor. Nat. Struct. Mol. Biol. 2005, 12, 935–936. [Google Scholar] [CrossRef] [PubMed]
- Shimi, T.; Koujin, T.; Segura-Totten, M.; Wilson, K.L.; Haraguchi, T.; Hiraoka, Y. Dynamic interaction between BAF and emerin revealed by FRAP, FLIP, and FRET analyses in living HeLa cells. J. Struct. Biol. 2004, 147, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Holaska, J.M.; Lee, K.K.; Kowalski, A.K.; Wilson, K.L. Transcriptional Repressor Germ Cell-less (GCL) and Barrier to Autointegration Factor (BAF) Compete for Binding to Emerin in Vitro. J. Biol. Chem. 2003, 278, 6969–6975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samson, C.; Petitalot, A.; Celli, F.; Herrada, I.; Ropars, V.; Le Du, M.H.; Nhiri, N.; Jacquet, E.; Arteni, A.A.; Buendia, B.; et al. Structural analysis of the ternary complex between lamin A/C, BAF and emerin identifies an interface disrupted in autosomal recessive progeroid diseases. Nucleic Acids Res. 2018, 46, 10460–10473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of A-Type Lamin Expression Compromises Nuclear Envelope Integrity Leading to Muscular Dystrophy. J. Cell Biol. 1999, 147, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Dubińska-Magiera, M.; Kozioł, K.; Machowska, M.; Piekarowicz, K.; Filipczak, D.; Rzepecki, R. Emerin Is Required for Proper Nucleus Reassembly after Mitosis: Implications for New Pathogenetic Mechanisms for Laminopathies Detected in EDMD1 Patients. Cells 2019, 8, 240. [Google Scholar] [CrossRef] [PubMed]
- Herrada, I.; Samson, C.; Velours, C.; Renault, L.; Östlund, C.; Chervy, P.; Puchkov, D.; Worman, H.J.; Buendia, B.; Zinn-Justin, S. Muscular Dystrophy Mutations Impair the Nuclear Envelope Emerin Self-assembly Properties. ACS Chem. Biol. 2015, 10, 2733–2742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, L.J.; Soshnev, A.A.; Geyer, P.K. Networking in the nucleus: A spotlight on LEM-domain proteins. Curr. Opin. Cell Biol. 2015, 34, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Ghirlando, R.; Lee, M.S.; Mizuuchi, K.; Krause, M.; Craigie, R. Barrier-to-autointegration factor (BAF) bridges DNA in a discrete, higher-order nucleoprotein complex. Proc. Natl. Acad. Sci. USA 2000, 97, 8997–9002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, F.; Mazzeo, D.; Patel, J.T.; Smallwood, D.T.; Ellis, J.A.; Shanahan, C.M.; Shackleton, S. Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J. Biol. Chem. 2010, 285, 3487–3498. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Jaalouk, D.E.; Vartiainen, M.K.; Lammerding, J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 2013, 497, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Manilal, S. Mutations in Emery-Dreifuss muscular dystrophy and their effects on emerin protein expression. Hum. Mol. Genet. 1998, 7, 855–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montes de Oca, R.; Shoemaker, C.J.; Gucek, M.; Cole, R.N.; Wilson, K.L. Barrier-to-Autointegration Factor Proteome Reveals Chromatin-Regulatory Partners. PLoS ONE 2009, 4, e7050. [Google Scholar] [CrossRef] [PubMed]
- Fairley, E.A.; Kendrick-Jones, J.; Ellis, J.A. The Emery-Dreifuss muscular dystrophy phenotype arises from aberrant targeting and binding of emerin at the inner nuclear membrane. J. Cell Sci. 1999, 112, 2571–2582. [Google Scholar]
- Mora, M.; Cartegni, L.; Di Blasi, C.; Barresi, R.; Bione, S.; Raffaele di Barletta, M.; Morandi, L.; Merlini, L.; Nigro, V.; Politano, L.; et al. X-linked emery-dreifuss muscular dystrophy can be diagnosed from skin biopsy or blood sample. Ann. Neurol. 1997, 42, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.W.; Bagshaw, J.; Aksmanovic, V.M.A.; Coomber, E.; McMahon, R.; Whittaker, J.L.; Morrison, P.J.; Kendrick-Jones, J.; Ellis, J.A. Genotype-phenotype analysis in X-linked Emery–Dreifuss muscular dystrophy and identification of a missense mutation associated with a milder phenotype. Neuromuscul. Disord. 1999, 9, 159–165. [Google Scholar] [CrossRef]
- Rudenskaia, G.E.; Tverskaia, S.M.; Chukhrova, A.L.; Zakliaz’minskaia, E.V.; Kuropatkina, I.V.; Dadali, E.L.; Perminov, V.S.; Poliakov, A.V. [Clinical, genealogical and molecular genetic study of Emery-Dreifuss muscular dystrophy]. Zh. Nevrol. Psikhiatr. Im S S Korsakova 2006, 106, 58–65. [Google Scholar] [PubMed]
- Harborth, J.; Elbashir, S.M.; Bechert, K.; Tuschl, T.; Weber, K. Identification of essential genes in cultured mammalian cells using small interfering RNAs. J. Cell Sci. 2001, 114, 4557–4565. [Google Scholar] [PubMed]
- Muchir, A.; van Engelen, B.G.; Lammens, M.; Mislow, J.M.; McNally, E.; Schwartz, K.; Bonne, G. Nuclear envelope alterations in fibroblasts from LGMD1B patients carrying nonsense Y259X heterozygous or homozygous mutation in lamin A/C gene. Exp. Cell Res. 2003, 291, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Lammerding, J.; Hsiao, J.; Schulze, P.C.; Kozlov, S.; Stewart, C.L.; Lee, R.T. Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J. Cell Biol. 2005, 170, 781–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maninová, M.; Vomastek, T. Dorsal stress fibers, transverse actin arcs, and perinuclear actin fibers form an interconnected network that induces nuclear movement in polarizing fibroblasts. FEBS J. 2016, 283, 3676–3693. [Google Scholar] [CrossRef] [Green Version]
- Versaevel, M.; Braquenier, J.-B.; Riaz, M.; Grevesse, T.; Lantoine, J.; Gabriele, S. Super-resolution microscopy reveals LINC complex recruitment at nuclear indentation sites. Sci. Rep. 2014, 4, 7362. [Google Scholar] [CrossRef]
- Thakar, K.; May, C.K.; Rogers, A.; Carroll, C.W. Opposing roles for distinct LINC complexes in regulation of the small GTPase RhoA. Mol. Biol. Cell 2017, 28, 182. [Google Scholar] [CrossRef]
- Lei, K.; Zhu, X.; Xu, R.; Shao, C.; Xu, T.; Zhuang, Y.; Han, M. Inner nuclear envelope proteins SUN1 and SUN2 play a prominent role in the DNA damage response. Curr. Biol. CB 2012, 22, 1609–1615. [Google Scholar] [CrossRef]
- Chen, X.; Li, W.; Chen, Y.; Li, X.; Li, H.; Huang, H.; Bu, F.; Pan, X.; Yang, Y.; Huang, C.; et al. Suppression of SUN2 by DNA methylation is associated with HSCs activation and hepatic fibrosis. Cell Death Dis. 2018, 9, 1021. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Reference | Host | Supplier |
|---|---|---|---|
| Anti-emerin | ab40688 | Rabbit | Abcam |
| Anti-lamin A + C [131C3] | ab8984 | Mouse | Abcam |
| Anti-SUN2 [EPR6557] | ab124916 | Rabbit | Abcam |
| Anti-BANF1/BAF [EPR7668] | ab129184 | Rabbit | Abcam |
| Anti-SUN1 | C3286 | Rabbit | Generously provided as a gift from D. Hodzic, Department of Ophthalmology and Visual Sciences, Washington University School of Medicine, St Louis, MO, USA |
| Gene | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| RPLPO | CTCCAAGCAGATGCAGCAGA | ATAGCCTTGCGCATCATGGT |
| EMD | CCCTGCCAGCCAGTCCCCTCG | CACCCCCACTGCTAAGGCAGTCAGC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Essawy, N.; Samson, C.; Petitalot, A.; Moog, S.; Bigot, A.; Herrada, I.; Marcelot, A.; Arteni, A.-A.; Coirault, C.; Zinn-Justin, S. An Emerin LEM-Domain Mutation Impairs Cell Response to Mechanical Stress. Cells 2019, 8, 570. https://doi.org/10.3390/cells8060570
Essawy N, Samson C, Petitalot A, Moog S, Bigot A, Herrada I, Marcelot A, Arteni A-A, Coirault C, Zinn-Justin S. An Emerin LEM-Domain Mutation Impairs Cell Response to Mechanical Stress. Cells. 2019; 8(6):570. https://doi.org/10.3390/cells8060570
Chicago/Turabian StyleEssawy, Nada, Camille Samson, Ambre Petitalot, Sophie Moog, Anne Bigot, Isaline Herrada, Agathe Marcelot, Ana-Andreea Arteni, Catherine Coirault, and Sophie Zinn-Justin. 2019. "An Emerin LEM-Domain Mutation Impairs Cell Response to Mechanical Stress" Cells 8, no. 6: 570. https://doi.org/10.3390/cells8060570