Melatonin and (−)-Epigallocatechin-3-Gallate: Partners in Fighting Cancer

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines and Cell Culture
2.3. MTT Assay
2.4. Cell Migration
2.5. Colony Formation
2.6. Western Blot
2.7. Preparation of Nuclear Fraction
2.8. Quinoprotein Measurement
2.9. Analysis of mRNA Expression by Real-Time PCR
2.10. Statistical Analysis
3. Results
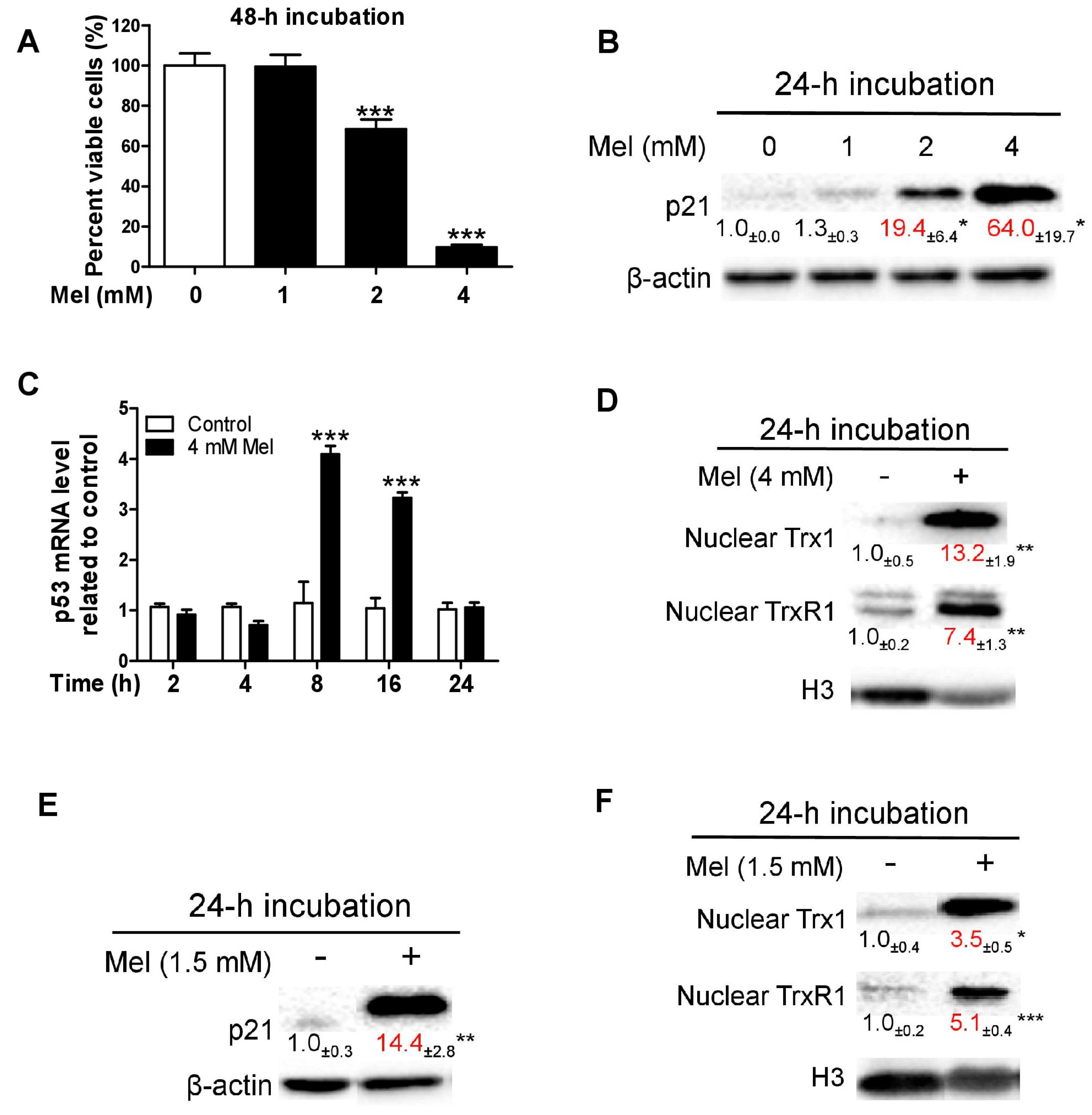
3.1. Melatonin-Induced p21 Correlates with the Increase of Nuclear Trx1 and TrxR1 in TCA8113 Cells
3.2. EGCG Increases Quinoprotein Levels in TCA8113 Cells
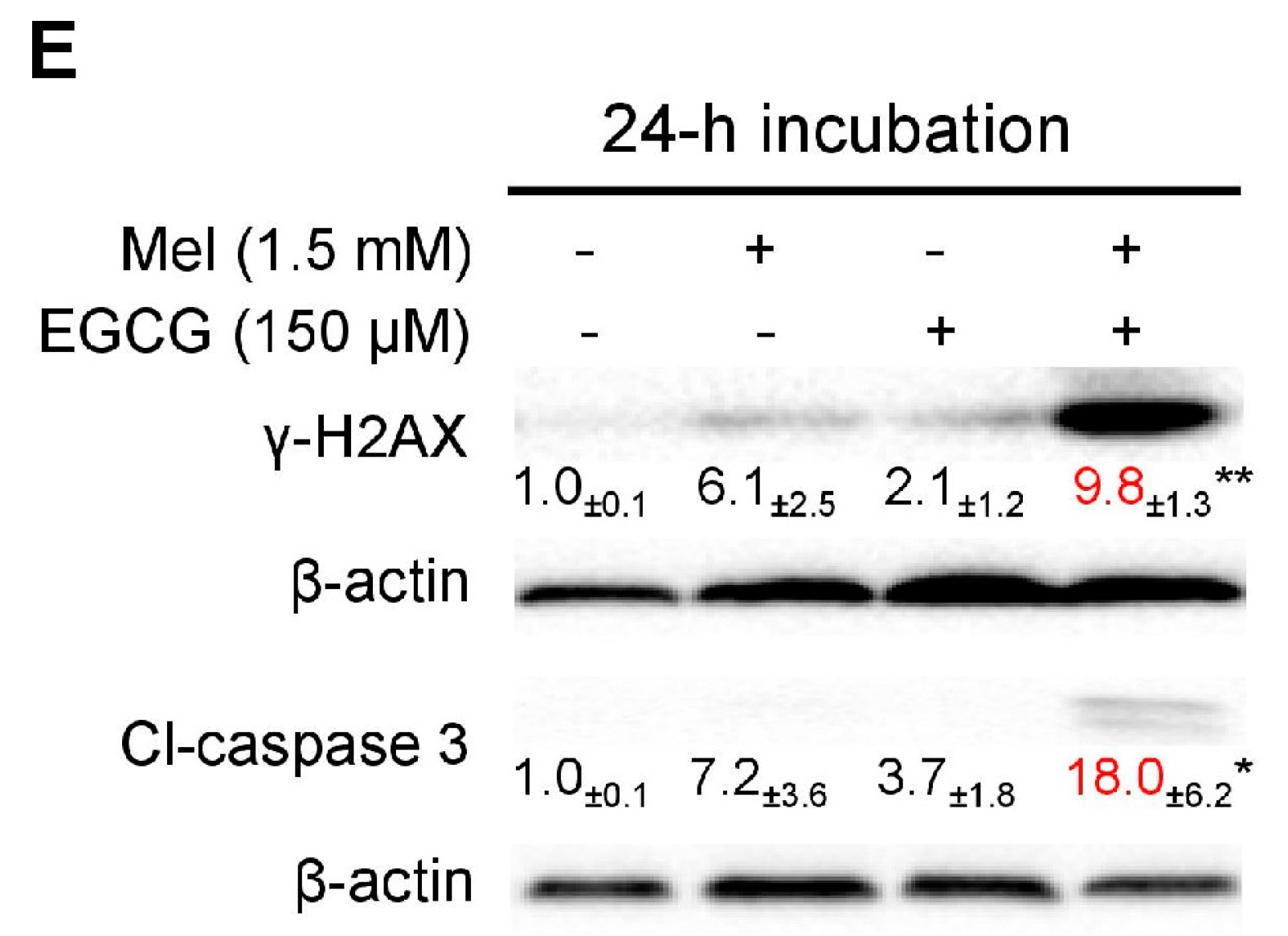
3.3. Concurrent Increases of p21 and Quinoprotein in TCA8113 Cells Treated with Melatonin and EGCG
3.4. Melatonin Reduces p21 in HepG2 Cells and Sensitizes the Cells to EGCG
3.5. EGCG Reverses Melatonin-Induced Pro-Survival Responses and Melatonin Does Not Impair Death-Promoting Action of EGCG in HepG2 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Maitra, S.; Bhattacharya, D.; Das, S.; Bhattacharya, S. Melatonin and its anti-glioma functions: A comprehensive review. Rev. Neurosci. 2019, 30, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Manchester, L.C.; Coto-Montes, A.; Boga, J.A.; Andersen, L.P.; Zhou, Z.; Galano, A.; Vriend, J.; Tan, D.X.; Reiter, R.J. Melatonin: An ancient molecule that makes oxygen metabolically tolerable. J. Pineal Res. 2015, 59, 403–419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.M.; Zhang, Y. Melatonin: A well-documented antioxidant with conditional pro-oxidant actions. J. Pineal Res. 2014, 57, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Rosales-Corral, S.A.; Tan, D.X.; Acuna-Castroviejo, D.; Qin, L.; Yang, S.F.; Xu, K. Melatonin, a Full Service Anti-Cancer Agent: Inhibition of Initiation, Progression and Metastasis. Int. J. Mol. Sci. 2017, 18, 843. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J. Mechanisms of cancer inhibition by melatonin. J. Pineal Res. 2004, 37, 213–214. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One molecule, many derivatives: A never-ending interaction of melatonin with reactive oxygen and nitrogen species? J. Pineal Res. 2007, 42, 28–42. [Google Scholar] [CrossRef]
- Rodriguez, C.; Mayo, J.C.; Sainz, R.M.; Antolin, I.; Herrera, F.; Martin, V.; Reiter, R.J. Regulation of antioxidant enzymes: A significant role for melatonin. J. Pineal Res. 2004, 36, 1–9. [Google Scholar] [CrossRef]
- Vriend, J.; Reiter, R.J. The Keap1-Nrf2-antioxidant response element pathway: A review of its regulation by melatonin and the proteasome. Mol. Cell Endocrinol. 2015, 401, 213–220. [Google Scholar] [CrossRef]
- Trachootham, D.; Zhou, Y.; Zhang, H.; Demizu, Y.; Chen, Z.; Pelicano, H.; Chiao, P.J.; Achanta, G.; Arlinghaus, R.B.; Liu, J.; et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. [Google Scholar] [CrossRef]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [PubMed]
- Raninga, P.V.; Trapani, G.D.; Tonissen, K.F. Cross Talk between Two Antioxidant Systems, Thioredoxin and DJ-1: Consequences for Cancer. Oncoscience 2014, 1, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A. Selenite in cancer therapy: A commentary on “Selenite induces apoptosis in sarcomatoid malignant mesothelioma cells through oxidative stress”. Free Radic. Biol. Med. 2006, 41, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Wu, S.; Wang, Y.; Wan, X.; Thompson, H.J.; Zhang, J. High-dose sodium selenite toxicity cannot be prevented by the co-administration of pharmacological levels of epigallocatechin-3-gallate which in turn aggravates the toxicity. Food Chem. Toxicol. 2013, 52, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Andersen, L.P.; Gogenur, I.; Rosenberg, J.; Reiter, R.J. The Safety of Melatonin in Humans. Clin. Drug Investig. 2016, 36, 169–175. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Sainz, R.M.; Mayo, J.C.; Lopez-Burillo, S. Melatonin: Reducing the toxicity and increasing the efficacy of drugs. J. Pharm. Pharmacol. 2002, 54, 1299–1321. [Google Scholar] [CrossRef] [PubMed]
- Majidinia, M.; Sadeghpour, A.; Mehrzadi, S.; Reiter, R.J.; Khatami, N.; Yousefi, B. Melatonin: A pleiotropic molecule that modulates DNA damage response and repair pathways. J. Pineal Res. 2017, 63, e12416. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.; Zhou, Y.; Meng, X.; Zhang, J.J.; Xu, D.P.; Li, H.B. Melatonin for the prevention and treatment of cancer. Oncotarget 2017, 8, 39896–39921. [Google Scholar] [CrossRef]
- Galley, H.F.; McCormick, B.; Wilson, K.L.; Lowes, D.A.; Colvin, L.; Torsney, C. Melatonin limits paclitaxel-induced mitochondrial dysfunction in vitro and protects against paclitaxel-induced neuropathic pain in the rat. J. Pineal Res. 2017, 63, e12444. [Google Scholar] [CrossRef]
- Pariente, R.; Pariente, J.A.; Rodriguez, A.B.; Espino, J. Melatonin sensitizes human cervical cancer HeLa cells to cisplatin-induced cytotoxicity and apoptosis: Effects on oxidative stress and DNA fragmentation. J. Pineal Res. 2016, 60, 55–64. [Google Scholar] [CrossRef]
- Manda, K.; Ueno, M.; Anzai, K. Cranial irradiation-induced inhibition of neurogenesis in hippocampal dentate gyrus of adult mice: Attenuation by melatonin pretreatment. J. Pineal Res. 2009, 46, 71–78. [Google Scholar] [CrossRef]
- Ortiz, F.; Acuna-Castroviejo, D.; Doerrier, C.; Dayoub, J.C.; Lopez, L.C.; Venegas, C.; Garcia, J.A.; Lopez, A.; Volt, H.; Luna-Sanchez, M.; et al. Melatonin blunts the mitochondrial/NLRP3 connection and protects against radiation-induced oral mucositis. J. Pineal Res. 2015, 58, 34–49. [Google Scholar] [CrossRef]
- Alonso-Gonzalez, C.; Gonzalez, A.; Martinez-Campa, C.; Gomez-Arozamena, J.; Cos, S. Melatonin sensitizes human breast cancer cells to ionizing radiation by downregulating proteins involved in double-strand DNA break repair. J. Pineal Res. 2015, 58, 189–197. [Google Scholar] [CrossRef]
- Alonso-Gonzalez, C.; Gonzalez, A.; Martinez-Campa, C.; Menendez-Menendez, J.; Gomez-Arozamena, J.; Garcia-Vidal, A.; Cos, S. Melatonin enhancement of the radiosensitivity of human breast cancer cells is associated with the modulation of proteins involved in estrogen biosynthesis. Cancer Lett. 2016, 370, 145–152. [Google Scholar] [CrossRef]
- Casado-Zapico, S.; Rodriguez-Blanco, J.; Garcia-Santos, G.; Martin, V.; Sanchez-Sanchez, A.M.; Antolin, I.; Rodriguez, C. Synergistic antitumor effect of melatonin with several chemotherapeutic drugs on human Ewing sarcoma cancer cells: Potentiation of the extrinsic apoptotic pathway. J. Pineal Res. 2010, 48, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Zhao, G.; Wang, Y.; Wang, D.; Sun, F.; Ning, J.; Wan, X.; Zhang, J. Safety and anti-hyperglycemic efficacy of various tea types in mice. Sci. Rep. 2016, 6, 31703. [Google Scholar] [CrossRef]
- Yang, C.S.; Hong, J. Prevention of chronic diseases by tea: Possible mechanisms and human relevance. Annu. Rev. Nutr. 2013, 33, 161–181. [Google Scholar] [CrossRef]
- Yang, C.S.; Wang, X.; Lu, G.; Picinich, S.C. Cancer prevention by tea: Animal studies, molecular mechanisms and human relevance. Nat. Rev. Cancer 2009, 9, 429–439. [Google Scholar] [CrossRef]
- Dong, R.; Wang, D.; Wang, X.; Zhang, K.; Chen, P.; Yang, C.S.; Zhang, J. Epigallocatechin-3-gallate enhances key enzymatic activities of hepatic thioredoxin and glutathione systems in selenium-optimal mice but activates hepatic Nrf2 responses in selenium-deficient mice. Redox Biol. 2016, 10, 221–232. [Google Scholar] [CrossRef]
- Wei, Y.; Chen, P.; Ling, T.; Wang, Y.; Dong, R.; Zhang, C.; Zhang, L.; Han, M.; Wang, D.; Wan, X.; et al. Certain (−)-epigallocatechin-3-gallate (EGCG) auto-oxidation products (EAOPs) retain the cytotoxic activities of EGCG. Food Chem. 2016, 204, 218–226. [Google Scholar] [CrossRef]
- Wang, D.; Taylor, E.W.; Wang, Y.; Wan, X.; Zhang, J. Encapsulated nanoepigallocatechin-3-gallate and elemental selenium nanoparticles as paradigms for nanochemoprevention. Int. J. Nanomed. 2012, 7, 1711–1721. [Google Scholar]
- Pae, M.; Ren, Z.; Meydani, M.; Shang, F.; Smith, D.; Meydani, S.N.; Wu, D. Dietary supplementation with high dose of epigallocatechin-3-gallate promotes inflammatory response in mice. J. Nutr. Biochem. 2012, 23, 526–531. [Google Scholar] [CrossRef]
- Shanafelt, T.D.; Call, T.G.; Zent, C.S.; LaPlant, B.; Bowen, D.A.; Roos, M.; Secreto, C.R.; Ghosh, A.K.; Kabat, B.F.; Lee, M.J.; et al. Phase I trial of daily oral Polyphenon E in patients with asymptomatic Rai stage 0 to II chronic lymphocytic leukemia. J. Clin. Oncol. 2009, 27, 3808–3814. [Google Scholar] [CrossRef]
- Bitzer, Z.T.; Elias, R.J.; Vijay-Kumar, M.; Lambert, J.D. (−)-epigallocatechin-3-gallate decreases colonic inflammation and permeability in a mouse model of colitis, but reduces macronutrient digestion and exacerbates weight loss. Mol. Nutr. Food Res. 2016, 60, 2267–2274. [Google Scholar] [CrossRef]
- Lambert, J.D.; Kennett, M.J.; Sang, S.; Reuhl, K.R.; Ju, J.; Yang, C.S. Hepatotoxicity of high oral dose (−)-epigallocatechin-3-gallate in mice. Food Chem. Toxicol. 2010, 48, 409–416. [Google Scholar] [CrossRef]
- James, K.D.; Forester, S.C.; Lambert, J.D. Dietary pretreatment with green tea polyphenol, (−)-epigallocatechin-3-gallate reduces the bioavailability and hepatotoxicity of subsequent oral bolus doses of (−)-epigallocatechin-3-gallate. Food Chem. Toxicol. 2015, 76, 103–108. [Google Scholar] [CrossRef]
- Mazzanti, G.; Menniti-Ippolito, F.; Moro, P.A.; Cassetti, F.; Raschetti, R.; Santuccio, C.; Mastrangelo, S. Hepatotoxicity from green tea: A review of the literature and two unpublished cases. Eur. J. Clin. Pharmacol. 2009, 65, 331–341. [Google Scholar] [CrossRef]
- Mazzanti, G.; Di Sotto, A.; Vitalone, A. Hepatotoxicity of green tea: An update. Arch. Toxicol. 2015, 89, 1175–1191. [Google Scholar] [CrossRef]
- Yates, A.A.; Erdman, J.W., Jr.; Shao, A.; Dolan, L.C.; Griffiths, J.C. Bioactive nutrients—Time for tolerable upper intake levels to address safety. Regul. Toxicol. Pharmacol. 2017, 84, 94–101. [Google Scholar] [CrossRef]
- Dekant, W.; Fujii, K.; Shibata, E.; Morita, O.; Shimotoyodome, A. Safety assessment of green tea based beverages and dried green tea extracts as nutritional supplements. Toxicol. Lett. 2017, 277, 104–108. [Google Scholar] [CrossRef]
- Hu, J.; Webster, D.; Cao, J.; Shao, A. The safety of green tea and green tea extract consumption in adults—Results of a systematic review. Regul. Toxicol. Pharmacol. 2018, 95, 412–433. [Google Scholar] [CrossRef]
- Setiawan, V.W.; Zhang, Z.F.; Yu, G.P.; Lu, Q.Y.; Li, Y.L.; Lu, M.L.; Wang, M.R.; Guo, C.H.; Yu, S.Z.; Kurtz, R.C.; et al. Protective effect of green tea on the risks of chronic gastritis and stomach cancer. Int. J. Cancer 2001, 92, 600–604. [Google Scholar] [CrossRef]
- Mu, L.N.; Lu, Q.Y.; Yu, S.Z.; Jiang, Q.W.; Cao, W.; You, N.C.; Setiawan, V.W.; Zhou, X.F.; Ding, B.G.; Wang, R.H.; et al. Green tea drinking and multigenetic index on the risk of stomach cancer in a Chinese population. Int. J. Cancer 2005, 116, 972–983. [Google Scholar] [CrossRef]
- Sasazuki, S.; Inoue, M.; Hanaoka, T.; Yamamoto, S.; Sobue, T.; Tsugane, S. Green tea consumption and subsequent risk of gastric cancer by subsite: The JPHC Study. Cancer Causes Control 2004, 15, 483–491. [Google Scholar] [CrossRef]
- Jian, L.; Xie, L.P.; Lee, A.H.; Binns, C.W. Protective effect of green tea against prostate cancer: A case-control study in southeast China. Int. J. Cancer 2004, 108, 130–135. [Google Scholar] [CrossRef]
- Kurahashi, N.; Sasazuki, S.; Iwasaki, M.; Inoue, M.; Tsugane, S.; Group, J.S. Green tea consumption and prostate cancer risk in Japanese men: A prospective study. Am. J. Epidemiol. 2008, 167, 71–77. [Google Scholar] [CrossRef]
- Yang, G.; Shu, X.O.; Li, H.; Chow, W.H.; Ji, B.T.; Zhang, X.; Gao, Y.T.; Zheng, W. Prospective cohort study of green tea consumption and colorectal cancer risk in women. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1219–1223. [Google Scholar] [CrossRef]
- Zhang, M.; Holman, C.D.; Huang, J.P.; Xie, X. Green tea and the prevention of breast cancer: A case-control study in Southeast China. Carcinogenesis 2007, 28, 1074–1078. [Google Scholar] [CrossRef]
- Zhong, L.; Goldberg, M.S.; Gao, Y.T.; Hanley, J.A.; Parent, M.E.; Jin, F. A population-based case-control study of lung cancer and green tea consumption among women living in Shanghai, China. Epidemiology 2001, 12, 695–700. [Google Scholar] [CrossRef]
- Wang, D.; Wei, Y.; Wang, T.; Wan, X.; Yang, C.S.; Reiter, R.J.; Zhang, J. Melatonin attenuates (−)-epigallocatehin-3-gallate-triggered hepatotoxicity without compromising its downregulation of hepatic gluconeogenic and lipogenic genes in mice. J. Pineal Res. 2015, 59, 497–507. [Google Scholar] [CrossRef]
- Wang, D.; Wang, Y.; Wan, X.; Yang, C.S.; Zhang, J. Green tea polyphenol (−)-epigallocatechin-3-gallate triggered hepatotoxicity in mice: Responses of major antioxidant enzymes and the Nrf2 rescue pathway. Toxicol. Appl. Pharmacol. 2015, 283, 65–74. [Google Scholar] [CrossRef]
- Zhang, K.; Dong, R.; Sun, K.; Wang, X.; Wang, J.; Yang, C.S.; Zhang, J. Synergistic toxicity of epigallocatechin-3-gallate and diethyldithiocarbamate, a lethal encounter involving redox-active copper. Free Radic. Biol. Med. 2017, 113, 143–156. [Google Scholar] [CrossRef]
- Ueno, M.; Masutani, H.; Arai, R.J.; Yamauchi, A.; Hirota, K.; Sakai, T.; Inamoto, T.; Yamaoka, Y.; Yodoi, J.; Nikaido, T. Thioredoxin-dependent redox regulation of p53-mediated p21 activation. J. Biol. Chem. 1999, 274, 35809–35815. [Google Scholar] [CrossRef]
- Ueno, M.; Matsutani, Y.; Nakamura, H.; Masutani, H.; Yagi, M.; Yamashiro, H.; Kato, H.; Inamoto, T.; Yamauchi, A.; Takahashi, R.; et al. Possible association of thioredoxin and p53 in breast cancer. Immunol. Lett. 2000, 75, 15–20. [Google Scholar] [CrossRef]
- Ishii, T.; Mori, T.; Tanaka, T.; Mizuno, D.; Yamaji, R.; Kumazawa, S.; Nakayama, T.; Akagawa, M. Covalent modification of proteins by green tea polyphenol (−)-epigallocatechin-3-gallate through autoxidation. Free Radic. Biol. Med. 2008, 45, 1384–1394. [Google Scholar] [CrossRef]
- Tao, L.; Park, J.Y.; Lambert, J.D. Differential prooxidative effects of the green tea polyphenol, (−)-epigallocatechin-3-gallate, in normal and oral cancer cells are related to differences in sirtuin 3 signaling. Mol. Nutr. Food Res. 2015, 59, 203–211. [Google Scholar] [CrossRef]
- Weiss, R.H. p21Waf1/Cip1 as a therapeutic target in breast and other cancers. Cancer Cell 2003, 4, 425–429. [Google Scholar] [CrossRef]
- Fan, S.; Chang, J.K.; Smith, M.L.; Duba, D.; Fornace, A.J., Jr.; O’Connor, P.M. Cells lacking CIP1/WAF1 genes exhibit preferential sensitivity to cisplatin and nitrogen mustard. Oncogene 1997, 14, 2127–2136. [Google Scholar] [CrossRef][Green Version]
- Lazzarini, R.; Moretti, S.; Orecchia, S.; Betta, P.G.; Procopio, A.; Catalano, A. Enhanced antitumor therapy by inhibition of p21waf1 in human malignant mesothelioma. Clin. Cancer Res. 2008, 14, 5099–5107. [Google Scholar] [CrossRef]
- Stewart, Z.A.; Mays, D.; Pietenpol, J.A. Defective G1-S cell cycle checkpoint function sensitizes cells to microtubule inhibitor-induced apoptosis. Cancer Res. 1999, 59, 3831–3837. [Google Scholar]
- Waldman, T.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Uncoupling of S phase and mitosis induced by anticancer agents in cells lacking p21. Nature 1996, 381, 713–716. [Google Scholar] [CrossRef]
- Li, Y.; Dowbenko, D.; Lasky, L.A. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J. Biol. Chem. 2002, 277, 11352–11361. [Google Scholar] [CrossRef]
- Cos, S.; Mediavilla, M.D.; Fernandez, R.; Gonzalez-Lamuno, D.; Sanchez-Barcelo, E.J. Does melatonin induce apoptosis in MCF-7 human breast cancer cells in vitro? J. Pineal Res. 2002, 32, 90–96. [Google Scholar] [CrossRef]
- Proietti, S.; Cucina, A.; Dobrowolny, G.; D’Anselmi, F.; Dinicola, S.; Masiello, M.G.; Pasqualato, A.; Palombo, A.; Morini, V.; Reiter, R.J.; et al. Melatonin down-regulates MDM2 gene expression and enhances p53 acetylation in MCF-7 cells. J. Pineal Res. 2014, 57, 120–129. [Google Scholar] [CrossRef]
- Arner, E.S. Focus on mammalian thioredoxin reductases--important selenoproteins with versatile functions. Biochim. Biophys. Acta 2009, 1790, 495–526. [Google Scholar] [CrossRef]
- Heilman, J.M.; Burke, T.J.; McClain, C.J.; Watson, W.H. Transactivation of gene expression by NF-kappaB is dependent on thioredoxin reductase activity. Free Radic. Biol. Med. 2011, 51, 1533–1542. [Google Scholar] [CrossRef][Green Version]
- Damdimopoulos, A.E.; Miranda-Vizuete, A.; Treuter, E.; Gustafsson, J.A.; Spyrou, G. An alternative splicing variant of the selenoprotein thioredoxin reductase is a modulator of estrogen signaling. J. Biol. Chem. 2004, 279, 38721–38729. [Google Scholar] [CrossRef]
- Yang, C.S.; Wang, H. Mechanistic issues concerning cancer prevention by tea catechins. Mol. Nutr. Food Res. 2011, 55, 819–831. [Google Scholar] [CrossRef]
- Forester, S.C.; Lambert, J.D. The role of antioxidant versus pro-oxidant effects of green tea polyphenols in cancer prevention. Mol. Nutr. Food Res. 2011, 55, 844–854. [Google Scholar] [CrossRef]
- Lambert, J.D.; Elias, R.J. The antioxidant and pro-oxidant activities of green tea polyphenols: A role in cancer prevention. Arch. Biochem. Biophys. 2010, 501, 65–72. [Google Scholar] [CrossRef]
- Martin, V.; Herrera, F.; Garcia-Santos, G.; Antolin, I.; Rodriguez-Blanco, J.; Medina, M.; Rodriguez, C. Involvement of protein kinase C in melatonin’s oncostatic effect in C6 glioma cells. J. Pineal Res. 2007, 43, 239–244. [Google Scholar] [CrossRef]
- Song, J.; Ma, S.J.; Luo, J.H.; Zhang, H.; Wang, R.X.; Liu, H.; Li, L.; Zhang, Z.G.; Zhou, R.X. Melatonin induces the apoptosis and inhibits the proliferation of human gastric cancer cells via blockade of the AKT/MDM2 pathway. Oncol. Rep. 2018, 39, 1975–1983. [Google Scholar] [CrossRef]
- Asada, M.; Yamada, T.; Ichijo, H.; Delia, D.; Miyazono, K.; Fukumuro, K.; Mizutani, S. Apoptosis inhibitory activity of cytoplasmic p21(Cip1/WAF1) in monocytic differentiation. EMBO J. 1999, 18, 1223–1234. [Google Scholar] [CrossRef]
- Fan, Y.; Borowsky, A.D.; Weiss, R.H. An antisense oligodeoxynucleotide to p21(Waf1/Cip1) causes apoptosis in human breast cancer cells. Mol. Cancer Ther. 2003, 2, 773–782. [Google Scholar]
- Weiss, R.H.; Marshall, D.; Howard, L.; Corbacho, A.M.; Cheung, A.T.; Sawai, E.T. Suppression of breast cancer growth and angiogenesis by an antisense oligodeoxynucleotide to p21(Waf1/Cip1). Cancer Lett. 2003, 189, 39–48. [Google Scholar] [CrossRef]
- Zhu, A.X. Systemic therapy of advanced hepatocellular carcinoma: How hopeful should we be? Oncologist 2006, 11, 790–800. [Google Scholar] [CrossRef]
- Niu, L.; Liu, L.; Yang, S.; Ren, J.; Lai, P.B.S.; Chen, G.G. New insights into sorafenib resistance in hepatocellular carcinoma: Responsible mechanisms and promising strategies. Biochim. Biophys. Acta 2017, 1868, 564–570. [Google Scholar] [CrossRef]
- Huynh, H.; Lee, J.W.; Chow, P.K.; Ngo, V.C.; Lew, G.B.; Lam, I.W.; Ong, H.S.; Chung, A.; Soo, K.C. Sorafenib induces growth suppression in mouse models of gastrointestinal stromal tumor. Mol. Cancer Ther. 2009, 8, 152–159. [Google Scholar] [CrossRef][Green Version]
- Inoue, H.; Hwang, S.H.; Wecksler, A.T.; Hammock, B.D.; Weiss, R.H. Sorafenib attenuates p21 in kidney cancer cells and augments cell death in combination with DNA-damaging chemotherapy. Cancer Biol. Ther. 2011, 12, 827–836. [Google Scholar] [CrossRef]
- Fan, L.; Sun, G.; Ma, T.; Zhong, F.; Lei, Y.; Li, X.; Wei, W. Melatonin reverses tunicamycin-induced endoplasmic reticulum stress in human hepatocellular carcinoma cells and improves cytotoxic response to doxorubicin by increasing CHOP and decreasing survivin. J. Pineal Res. 2013, 55, 184–194. [Google Scholar] [CrossRef]
- Prieto-Dominguez, N.; Ordonez, R.; Fernandez, A.; Mendez-Blanco, C.; Baulies, A.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Mauriz, J.L.; Gonzalez-Gallego, J. Melatonin-induced increase in sensitivity of human hepatocellular carcinoma cells to sorafenib is associated with reactive oxygen species production and mitophagy. J. Pineal Res. 2016, 61, 396–407. [Google Scholar] [CrossRef]
- Lin, S.; Hoffmann, K.; Gao, C.; Petrulionis, M.; Herr, I.; Schemmer, P. Melatonin promotes sorafenib-induced apoptosis through synergistic activation of JNK/c-jun pathway in human hepatocellular carcinoma. J. Pineal Res. 2017, 62, e12398. [Google Scholar] [CrossRef]
- Zha, L.; Fan, L.; Sun, G.; Wang, H.; Ma, T.; Zhong, F.; Wei, W. Melatonin sensitizes human hepatoma cells to endoplasmic reticulum stress-induced apoptosis. J. Pineal Res. 2012, 52, 322–331. [Google Scholar] [CrossRef]
- Hao, J.; Li, Z.; Zhang, C.; Yu, W.; Tang, Z.; Li, Y.; Feng, X.; Gao, Y.; Liu, Q.; Huang, W.; et al. Targeting NF-kappaB/AP-2beta signaling to enhance antitumor activity of cisplatin by melatonin in hepatocellular carcinoma cells. Am. J. Cancer Res. 2017, 7, 13–27. [Google Scholar]
- Mortezaee, K. Human hepatocellular carcinoma: Protection by melatonin. J. Cell. Physiol. 2018, 233, 6486–6508. [Google Scholar] [CrossRef]
- Martin-Renedo, J.; Mauriz, J.L.; Jorquera, F.; Ruiz-Andres, O.; Gonzalez, P.; Gonzalez-Gallego, J. Melatonin induces cell cycle arrest and apoptosis in hepatocarcinoma HepG2 cell line. J. Pineal Res. 2008, 45, 532–540. [Google Scholar] [CrossRef]
- Carbajo-Pescador, S.; Martin-Renedo, J.; Garcia-Palomo, A.; Tunon, M.J.; Mauriz, J.L.; Gonzalez-Gallego, J. Changes in the expression of melatonin receptors induced by melatonin treatment in hepatocarcinoma HepG2 cells. J. Pineal Res. 2009, 47, 330–338. [Google Scholar] [CrossRef]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; He, Y.; Wu, X.; Zhao, G.; Zhang, K.; Yang, C.S.; Reiter, R.J.; Zhang, J. Melatonin and (−)-Epigallocatechin-3-Gallate: Partners in Fighting Cancer. Cells 2019, 8, 745. https://doi.org/10.3390/cells8070745
Zhang L, He Y, Wu X, Zhao G, Zhang K, Yang CS, Reiter RJ, Zhang J. Melatonin and (−)-Epigallocatechin-3-Gallate: Partners in Fighting Cancer. Cells. 2019; 8(7):745. https://doi.org/10.3390/cells8070745
Chicago/Turabian StyleZhang, Lingyun, Yufeng He, Ximing Wu, Guangshan Zhao, Ke Zhang, Chung S. Yang, Russel J. Reiter, and Jinsong Zhang. 2019. "Melatonin and (−)-Epigallocatechin-3-Gallate: Partners in Fighting Cancer" Cells 8, no. 7: 745. https://doi.org/10.3390/cells8070745
APA StyleZhang, L., He, Y., Wu, X., Zhao, G., Zhang, K., Yang, C. S., Reiter, R. J., & Zhang, J. (2019). Melatonin and (−)-Epigallocatechin-3-Gallate: Partners in Fighting Cancer. Cells, 8(7), 745. https://doi.org/10.3390/cells8070745