Interplay between Intrinsic and Innate Immunity during HIV Infection

Abstract
:1. Introduction
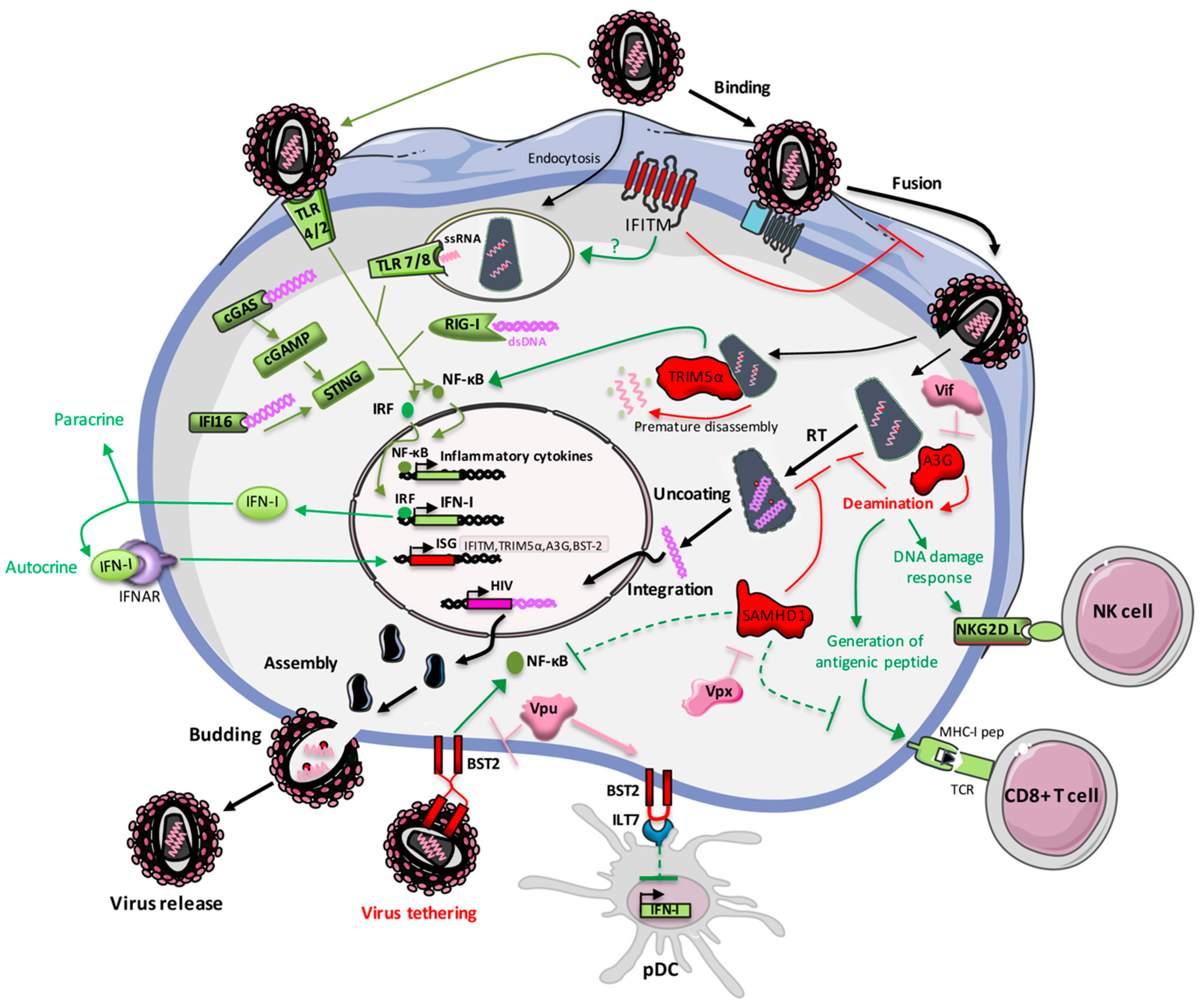
2. Activation of Innate Immunity Following HIV Sensing
2.1. General Overview of Pattern Recognition Receptors Signaling after Pathogen Associated Molecular Patterns Recognition
2.2. HIV Sensing by Pattern Recognition Receptors
2.2.1. Sensing by Toll-Like Receptors
2.2.2. Sensing by Cytoplasmic Receptors
2.2.3. Activation of the Inflammasome
3. Restriction Factors Belonging to the Interferon Stimulated Genes Family
3.1. IFITM
3.2. TRIM5α
3.3. APOBEC3G
3.4. SAMHD1
3.5. Mx2
3.6. Tetherin/BST-2
3.7. Other Interferon Stimulated Genes Restricting HIV Infection
3.7.1. Cholesterol-25-Hydroxylase
3.7.2. Zinc-Finger Antiviral Protein (ZAP)
3.7.3. Schlafen 11
3.7.4. ISG15
3.7.5. Guanylate-Binding Protein 5 (GBP5)
4. Restriction Factors Shaping Immunity
4.1. Restriction Factors Promoting Immunity
4.1.1. IFITM
4.1.2. TRIM5α
4.1.3. APOBEC3G
4.1.4. BST-2
4.1.5. Zinc-Finger Antiviral Protein (ZAP)
4.1.6. Guanylate-Binding Protein 5 (GBP5)
4.2. Restriction Factors Negatively Regulating Immunity
4.2.1. SAMHD1
4.2.2. BST-2
4.2.3. Cholesterol-25-Hydroxylase
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- D’Urbano, V.; De Crignis, E.; Re, M.C. Host restriction factors and human immunodeficiency virus (HIV-1): A dynamic interplay involving all phases of the viral life cycle. Curr. HIV Res. 2018, 16, 184–207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, B.; Pomerantz, R.J.; Zhang, C.; Arunachalam, S.C.; Gao, L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 2003, 424, 94–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Ségéral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [Green Version]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Pan, Q.; Rong, L.; He, W.; Liu, S.-L.; Liang, C. The IFITM proteins inhibit HIV-1 infection. J. Virol. 2011, 85, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Goujon, C.; Moncorgé, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hué, S.; Barclay, W.S.; Schulz, R.; Malim, M.H. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Kao, E.; Gao, X.; Sandig, H.; Limmer, K.; Pavon-Eternod, M.; Jones, T.E.; Landry, S.; Pan, T.; Weitzman, M.D.; et al. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature 2012, 491, 125–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadler, A.J.; Williams, B.R.G. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef]
- van Montfoort, N.; Olagnier, D.; Hiscott, J. Unmasking immune sensing of retroviruses: Interplay between innate sensors and host effectors. Cytokine Growth Factor Rev. 2014, 25, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Colomer-Lluch, M.; Ruiz, A.; Moris, A.; Prado, J.G. Restriction Factors: From Intrinsic Viral Restriction to Shaping Cellular Immunity Against HIV-1. Front. Immunol. 2018, 9, 2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A.J.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef]
- Medzhitov, R.; Janeway, C. The Toll receptor family and microbial recognition. Trends Microbiol. 2000, 8, 452–456. [Google Scholar] [CrossRef]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef]
- Chen, K.; Liu, J.; Cao, X. Regulation of type I interferon signaling in immunity and inflammation: A comprehensive review. J. Autoimmun. 2017, 83, 1–11. [Google Scholar] [CrossRef]
- Nazli, A.; Kafka, J.K.; Ferreira, V.H.; Anipindi, V.; Mueller, K.; Osborne, B.J.; Dizzell, S.; Chauvin, S.; Mian, M.F.; Ouellet, M.; et al. HIV-1 gp120 induces TLR2- and TLR4-mediated innate immune activation in human female genital epithelium. J. Immunol. 2013, 191, 4246–4258. [Google Scholar] [CrossRef]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef]
- Beignon, A.-S.; McKenna, K.; Skoberne, M.; Manches, O.; DaSilva, I.; Kavanagh, D.G.; Larsson, M.; Gorelick, R.J.; Lifson, J.D.; Bhardwaj, N. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Invest. 2005, 115, 3265–3275. [Google Scholar] [CrossRef]
- Fonteneau, J.-F.; Larsson, M.; Beignon, A.-S.; McKenna, K.; Dasilva, I.; Amara, A.; Liu, Y.-J.; Lifson, J.D.; Littman, D.R.; Bhardwaj, N. Human immunodeficiency virus type 1 activates plasmacytoid dendritic cells and concomitantly induces the bystander maturation of myeloid dendritic cells. J. Virol. 2004, 78, 5223–5232. [Google Scholar] [CrossRef]
- Meier, A.; Alter, G.; Frahm, N.; Sidhu, H.; Li, B.; Bagchi, A.; Teigen, N.; Streeck, H.; Stellbrink, H.-J.; Hellman, J.; et al. MyD88-dependent immune activation mediated by human immunodeficiency virus type 1-encoded Toll-like receptor ligands. J. Virol. 2007, 81, 8180–8191. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Lindsay, R.J.; Kulkarni, S.; Lifson, J.D.; Carrington, M.; Altfeld, M. Polymorphisms in interferon regulatory factor 7 reduce interferon-α responses of plasmacytoid dendritic cells to HIV-1. AIDS 2011, 25, 715–717. [Google Scholar] [CrossRef]
- Gringhuis, S.I.; van der Vlist, M.; van den Berg, L.M.; den Dunnen, J.; Litjens, M.; Geijtenbeek, T.B.H. HIV-1 exploits innate signaling by TLR8 and DC-SIGN for productive infection of dendritic cells. Nat. Immunol. 2010, 11, 419–426. [Google Scholar] [CrossRef]
- Cingöz, O.; Goff, S.P. HIV-1 is a poor inducer of innate immune responses. mBio 2019, 10. [Google Scholar] [CrossRef]
- Martinson, J.A.; Roman-Gonzalez, A.; Tenorio, A.R.; Montoya, C.J.; Gichinga, C.N.; Rugeles, M.T.; Tomai, M.; Krieg, A.M.; Ghanekar, S.; Baum, L.L.; et al. Dendritic cells from HIV-1 infected individuals are less responsive to toll-like receptor (TLR) ligands. Cell. Immunol. 2007, 250, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Blanchet, F.P.; Moris, A.; Nikolic, D.S.; Lehmann, M.; Cardinaud, S.; Stalder, R.; Garcia, E.; Dinkins, C.; Leuba, F.; Wu, L.; et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 2010, 32, 654–669. [Google Scholar] [CrossRef]
- Henrick, B.M.; Yao, X.-D.; Zahoor, M.A.; Abimiku, A.; Osawe, S.; Rosenthal, K.L. TLR10 senses HIV-1 proteins and significantly enhances HIV-1 infection. Front. Immunol. 2019, 10, 482. [Google Scholar] [CrossRef]
- Berg, R.K.; Melchjorsen, J.; Rintahaka, J.; Diget, E.; Søby, S.; Horan, K.A.; Gorelick, R.J.; Matikainen, S.; Larsen, C.S.; Ostergaard, L.; et al. Genomic HIV RNA induces innate immune responses through RIG-I-dependent sensing of secondary-structured RNA. PLoS ONE 2012, 7, e29291. [Google Scholar] [CrossRef]
- Solis, M.; Nakhaei, P.; Jalalirad, M.; Lacoste, J.; Douville, R.; Arguello, M.; Zhao, T.; Laughrea, M.; Wainberg, M.A.; Hiscott, J. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J. Virol. 2011, 85, 1224–1236. [Google Scholar] [CrossRef]
- Nasr, N.; Alshehri, A.A.; Wright, T.K.; Shahid, M.; Heiner, B.M.; Harman, A.N.; Botting, R.A.; Helbig, K.J.; Beard, M.R.; Suzuki, K.; et al. Mechanism of interferon-stimulated gene induction in HIV-1-infected macrophages. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, H.; Wu, J.; Zhang, X.; Sun, L.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 2013, 51, 226–235. [Google Scholar] [CrossRef]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.-P.; Ludwig, J.; Hornung, V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef]
- Ablasser, A.; Schmid-Burgk, J.L.; Hemmerling, I.; Horvath, G.L.; Schmidt, T.; Latz, E.; Hornung, V. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 2013, 503, 530–534. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Wu, J.; Wu, Y.-T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef]
- Manel, N.; Hogstad, B.; Wang, Y.; Levy, D.E.; Unutmaz, D.; Littman, D.R. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature 2010, 467, 214–217. [Google Scholar] [CrossRef]
- Peng, K.; Muranyi, W.; Glass, B.; Laketa, V.; Yant, S.R.; Tsai, L.; Cihlar, T.; Müller, B.; Kräusslich, H.-G. Quantitative microscopy of functional HIV post-entry complexes reveals association of replication with the viral capsid. Elife 2014, 3, e04114. [Google Scholar] [CrossRef]
- Chin, C.R.; Perreira, J.M.; Savidis, G.; Portmann, J.M.; Aker, A.M.; Feeley, E.M.; Smith, M.C.; Brass, A.L. Direct visualization of HIV-1 replication intermediates shows that capsid and CPSF6 modulate HIV-1 intra-nuclear invasion and integration. Cell Rep. 2015, 13, 1717–1731. [Google Scholar] [CrossRef]
- Lahaye, X.; Satoh, T.; Gentili, M.; Cerboni, S.; Conrad, C.; Hurbain, I.; El Marjou, A.; Lacabaratz, C.; Lelièvre, J.-D.; Manel, N. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity 2013, 39, 1132–1142. [Google Scholar] [CrossRef]
- Lahaye, X.; Gentili, M.; Silvin, A.; Conrad, C.; Picard, L.; Jouve, M.; Zueva, E.; Maurin, M.; Nadalin, F.; Knott, G.J.; et al. NONO detects the nuclear HIV capsid to promote cGAS-mediated innate immune activation. Cell 2018, 175, 488–501.e22. [Google Scholar] [CrossRef]
- Jakobsen, M.R.; Bak, R.O.; Andersen, A.; Berg, R.K.; Jensen, S.B.; Tengchuan, J.; Jin, T.; Laustsen, A.; Hansen, K.; Ostergaard, L.; et al. IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc. Natl. Acad. Sci. USA 2013, 110, E4571–E4580. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Gao, J.; Taxman, D.J.; Ting, J.P.Y.; Su, L. HIV-1 infection induces interleukin-1β production via TLR8 protein-dependent and NLRP3 inflammasome mechanisms in human monocytes. J. Biol. Chem. 2014, 289, 21716–21726. [Google Scholar] [CrossRef]
- Chattergoon, M.A.; Latanich, R.; Quinn, J.; Winter, M.E.; Buckheit, R.W.; Blankson, J.N.; Pardoll, D.; Cox, A.L. HIV and HCV activate the inflammasome in monocytes and macrophages via endosomal Toll-like receptors without induction of type 1 interferon. PLoS Pathog. 2014, 10, e1004082. [Google Scholar] [CrossRef]
- Feria, M.G.; Taborda, N.A.; Hernandez, J.C.; Rugeles, M.T. HIV replication is associated to inflammasomes activation, IL-1β, IL-18 and caspase-1 expression in GALT and peripheral blood. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Bailey, C.C.; Kondur, H.R.; Huang, I.-C.; Farzan, M. Interferon-induced transmembrane protein 3 is a type II transmembrane protein. J. Biol. Chem. 2013, 288, 32184–32193. [Google Scholar] [CrossRef]
- Brass, A.L.; Huang, I.-C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef]
- Jiang, D.; Weidner, J.M.; Qing, M.; Pan, X.-B.; Guo, H.; Xu, C.; Zhang, X.; Birk, A.; Chang, J.; Shi, P.-Y.; et al. Identification of five interferon-induced cellular proteins that inhibit west nile virus and dengue virus infections. J. Virol. 2010, 84, 8332–8341. [Google Scholar] [CrossRef]
- Savidis, G.; Perreira, J.M.; Portmann, J.M.; Meraner, P.; Guo, Z.; Green, S.; Brass, A.L. The IFITMs inhibit zika virus replication. Cell Rep. 2016, 15, 2323–2330. [Google Scholar] [CrossRef]
- Wilkins, C.; Woodward, J.; Lau, D.T.-Y.; Barnes, A.; Joyce, M.; McFarlane, N.; McKeating, J.A.; Tyrrell, D.L.; Gale, M. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology 2013, 57, 461–469. [Google Scholar] [CrossRef]
- Huang, I.-C.; Bailey, C.C.; Weyer, J.L.; Radoshitzky, S.R.; Becker, M.M.; Chiang, J.J.; Brass, A.L.; Ahmed, A.A.; Chi, X.; Dong, L.; et al. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog. 2011, 7, e1001258. [Google Scholar] [CrossRef]
- Wrensch, F.; Karsten, C.B.; Gnirß, K.; Hoffmann, M.; Lu, K.; Takada, A.; Winkler, M.; Simmons, G.; Pöhlmann, S. Interferon-induced transmembrane protein-mediated inhibition of host cell entry of ebolaviruses. J. Infect. Dis. 2015, 212, S210–S218. [Google Scholar] [CrossRef]
- Mudhasani, R.; Tran, J.P.; Retterer, C.; Radoshitzky, S.R.; Kota, K.P.; Altamura, L.A.; Smith, J.M.; Packard, B.Z.; Kuhn, J.H.; Costantino, J.; et al. IFITM-2 and IFITM-3 but not IFITM-1 restrict Rift Valley fever virus. J. Virol. 2013, 87, 8451–8464. [Google Scholar] [CrossRef]
- Zheng, X.-Y.; Bian, P.-Y.; Ye, C.-T.; Ye, W.; Ma, H.-W.; Tang, K.; Zhang, C.-M.; Lei, Y.-F.; Wei, X.; Wang, P.-Z.; et al. Interferon-induced transmembrane protein 3 inhibits hantaan virus infection, and its single nucleotide polymorphism rs12252 influences the severity of hemorrhagic fever with renal syndrome. Front. Immunol. 2016, 7, 535. [Google Scholar] [CrossRef]
- Anafu, A.A.; Bowen, C.H.; Chin, C.R.; Brass, A.L.; Holm, G.H. Interferon-inducible transmembrane protein 3 (IFITM3) restricts reovirus cell entry. J. Biol. Chem. 2013, 288, 17261–17271. [Google Scholar] [CrossRef]
- Li, C.; Du, S.; Tian, M.; Wang, Y.; Bai, J.; Tan, P.; Liu, W.; Yin, R.; Wang, M.; Jiang, Y.; et al. The host restriction factor interferon-inducible transmembrane protein 3 inhibits vaccinia virus infection. Front. Immunol. 2018, 9, 228. [Google Scholar] [CrossRef]
- Smith, S.E.; Busse, D.C.; Binter, S.; Weston, S.; Diaz Soria, C.; Laksono, B.M.; Clare, S.; Van Nieuwkoop, S.; Van den Hoogen, B.G.; Clement, M.; et al. Interferon-induced transmembrane protein 1 restricts replication of viruses that enter cells via the plasma membrane. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Li, K.; Jia, R.; Li, M.; Zheng, Y.-M.; Miao, C.; Yao, Y.; Ji, H.-L.; Geng, Y.; Qiao, W.; Albritton, L.M.; et al. A sorting signal suppresses IFITM1 restriction of viral entry. J. Biol. Chem. 2015, 290, 4248–4259. [Google Scholar] [CrossRef]
- Jia, R.; Xu, F.; Qian, J.; Yao, Y.; Miao, C.; Zheng, Y.-M.; Liu, S.-L.; Guo, F.; Geng, Y.; Qiao, W.; et al. Identification of an endocytic signal essential for the antiviral action of IFITM3. Cell. Microbiol. 2014, 16, 1080–1093. [Google Scholar] [CrossRef]
- Li, K.; Markosyan, R.M.; Zheng, Y.-M.; Golfetto, O.; Bungart, B.; Li, M.; Ding, S.; He, Y.; Liang, C.; Lee, J.C.; et al. IFITM proteins restrict viral membrane hemifusion. PLoS Pathog. 2013, 9, e1003124. [Google Scholar] [CrossRef]
- Weidner, J.M.; Jiang, D.; Pan, X.-B.; Chang, J.; Block, T.M.; Guo, J.-T. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J. Virol. 2010, 84, 12646–12657. [Google Scholar] [CrossRef]
- Compton, A.A.; Bruel, T.; Porrot, F.; Mallet, A.; Sachse, M.; Euvrard, M.; Liang, C.; Casartelli, N.; Schwartz, O. IFITM proteins incorporated into HIV-1 virions impair viral fusion and spread. Cell Host Microbe 2014, 16, 736–747. [Google Scholar] [CrossRef]
- Desai, T.M.; Marin, M.; Chin, C.R.; Savidis, G.; Brass, A.L.; Melikyan, G.B. IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog. 2014, 10, e1004048. [Google Scholar] [CrossRef]
- Spence, J.S.; He, R.; Hoffmann, H.-H.; Das, T.; Thinon, E.; Rice, C.M.; Peng, T.; Chandran, K.; Hang, H.C. IFITM3 directly engages and shuttles incoming virus particles to lysosomes. Nat. Chem. Biol. 2019, 15, 259–268. [Google Scholar] [CrossRef]
- Tartour, K.; Appourchaux, R.; Gaillard, J.; Nguyen, X.-N.; Durand, S.; Turpin, J.; Beaumont, E.; Roch, E.; Berger, G.; Mahieux, R.; et al. IFITM proteins are incorporated onto HIV-1 virion particles and negatively imprint their infectivity. Retrovirology 2014, 11, 103. [Google Scholar] [CrossRef]
- Yu, J.; Li, M.; Wilkins, J.; Ding, S.; Swartz, T.H.; Esposito, A.M.; Zheng, Y.-M.; Freed, E.O.; Liang, C.; Chen, B.K.; et al. IFITM proteins restrict HIV-1 infection by antagonizing the envelope glycoprotein. Cell Rep. 2015, 13, 145–156. [Google Scholar] [CrossRef]
- Tartour, K.; Nguyen, X.-N.; Appourchaux, R.; Assil, S.; Barateau, V.; Bloyet, L.-M.; Burlaud Gaillard, J.; Confort, M.-P.; Escudero-Perez, B.; Gruffat, H.; et al. Interference with the production of infectious viral particles and bimodal inhibition of replication are broadly conserved antiviral properties of IFITMs. PLoS Pathog. 2017, 13, e1006610. [Google Scholar] [CrossRef]
- Appourchaux, R.; Delpeuch, M.; Zhong, L.; Burlaud-Gaillard, J.; Tartour, K.; Savidis, G.; Brass, A.; Etienne, L.; Roingeard, P.; Cimarelli, A. Functional mapping of regions involved in the negative imprinting of virion particle infectivity and in target cell protection by interferon-induced transmembrane protein 3 against HIV-1. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Jaffe, E.A.; Armellino, D.; Lam, G.; Cordon-Cardo, C.; Murray, H.W.; Evans, R.L. IFN-gamma and IFN-alpha induce the expression and synthesis of Leu 13 antigen by cultured human endothelial cells. J. Immunol. 1989, 143, 3961–3966. [Google Scholar]
- Siegrist, F.; Ebeling, M.; Certa, U. The small interferon-induced transmembrane genes and proteins. J. Interferon Cytokine Res. 2011, 31, 183–197. [Google Scholar] [CrossRef]
- Allen, E.K.; Randolph, A.G.; Bhangale, T.; Dogra, P.; Ohlson, M.; Oshansky, C.M.; Zamora, A.E.; Shannon, J.P.; Finkelstein, D.; Dressen, A.; et al. SNP-mediated disruption of CTCF binding at the IFITM3 promoter is associated with risk of severe influenza in humans. Nat. Med. 2017, 23, 975–983. [Google Scholar] [CrossRef]
- Diamond, M.S.; Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2013, 13, 46–57. [Google Scholar] [CrossRef]
- Zhao, X.; Li, J.; Winkler, C.A.; An, P.; Guo, J.-T. IFITM genes, variants, and their roles in the control and pathogenesis of viral infections. Front. Microbiol. 2018, 9, 3228. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, J.; Li, M.; Yang, H.; Zhang, C. Evolutionary dynamics of the interferon-induced transmembrane gene family in vertebrates. PLoS ONE 2012, 7, e49265. [Google Scholar] [CrossRef]
- Ozato, K.; Shin, D.-M.; Chang, T.-H.; Morse, H.C. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, S. TRIM family proteins: Roles in autophagy, immunity, and carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef]
- Rajsbaum, R.; García-Sastre, A.; Versteeg, G.A. TRIMmunity: The roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J. Mol. Biol. 2014, 426, 1265–1284. [Google Scholar] [CrossRef]
- Nisole, S.; Stoye, J.P.; Saïb, A. TRIM family proteins: Retroviral restriction and antiviral defence. Nat. Rev. Microbiol. 2005, 3, 799–808. [Google Scholar] [CrossRef]
- Goff, S.P. Retrovirus restriction factors. Mol. Cell 2004, 16, 849–859. [Google Scholar] [CrossRef]
- Hatziioannou, T.; Perez-Caballero, D.; Yang, A.; Cowan, S.; Bieniasz, P.D. Retrovirus resistance factors Ref1 and Lv1 are species-specific variants of TRIM5alpha. Proc. Natl. Acad. Sci. USA 2004, 101, 10774–10779. [Google Scholar] [CrossRef]
- Keckesova, Z.; Ylinen, L.M.J.; Towers, G.J. The human and African green monkey TRIM5alpha genes encode Ref1 and Lv1 retroviral restriction factor activities. Proc. Natl. Acad. Sci. USA 2004, 101, 10780–10785. [Google Scholar] [CrossRef]
- Perron, M.J.; Stremlau, M.; Song, B.; Ulm, W.; Mulligan, R.C.; Sodroski, J. TRIM5alpha mediates the postentry block to N-tropic murine leukemia viruses in human cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11827–11832. [Google Scholar] [CrossRef]
- Yap, M.W.; Nisole, S.; Lynch, C.; Stoye, J.P. Trim5alpha protein restricts both HIV-1 and murine leukemia virus. Proc. Natl. Acad. Sci. USA 2004, 101, 10786–10791. [Google Scholar] [CrossRef]
- Grütter, M.G.; Luban, J. TRIM5 structure, HIV-1 capsid recognition, and innate immune signaling. Curr. Opin. Virol. 2012, 2, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Pertel, T.; Hausmann, S.; Morger, D.; Züger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, E.E.; Miyoshi, H.; Nagai, Y.; Shioda, T. A specific region of 37 amino acid residues in the SPRY (B30.2) domain of African green monkey TRIM5alpha determines species-specific restriction of simian immunodeficiency virus SIVmac infection. J. Virol. 2005, 79, 8870–8877. [Google Scholar] [CrossRef]
- Towers, G.; Bock, M.; Martin, S.; Takeuchi, Y.; Stoye, J.P.; Danos, O. A conserved mechanism of retrovirus restriction in mammals. Proc. Natl. Acad. Sci. USA 2000, 97, 12295–12299. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, C.M.S.; Sarrami-Forooshani, R.; Setiawan, L.C.; Zijlstra-Willems, E.M.; van Hamme, J.L.; Tigchelaar, W.; van der Wel, N.N.; Kootstra, N.A.; Gringhuis, S.I.; Geijtenbeek, T.B.H. Receptor usage dictates HIV-1 restriction by human TRIM5α in dendritic cell subsets. Nature 2016, 540, 448–452. [Google Scholar] [CrossRef]
- Li, Y.-L.; Chandrasekaran, V.; Carter, S.D.; Woodward, C.L.; Christensen, D.E.; Dryden, K.A.; Pornillos, O.; Yeager, M.; Ganser-Pornillos, B.K.; Jensen, G.J.; et al. Primate TRIM5 proteins form hexagonal nets on HIV-1 capsids. Elife 2016, 5. [Google Scholar] [CrossRef]
- Yang, Y.; Brandariz-Nuñez, A.; Fricke, T.; Ivanov, D.N.; Sarnak, Z.; Diaz-Griffero, F. Binding of the rhesus TRIM5α PRYSPRY domain to capsid is necessary but not sufficient for HIV-1 restriction. Virology 2014, 448, 217–228. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.K.; Chandrasekaran, V.; Pornillos, O.; Sodroski, J.G.; Sundquist, W.I.; Yeager, M. Hexagonal assembly of a restricting TRIM5alpha protein. Proc. Natl. Acad. Sci. USA 2011, 108, 534–539. [Google Scholar] [CrossRef]
- Yap, M.W.; Nisole, S.; Stoye, J.P. A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr. Biol. 2005, 15, 73–78. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Stremlau, M.; Lee, M.; Sodroski, J. Removal of arginine 332 allows human TRIM5alpha to bind human immunodeficiency virus capsids and to restrict infection. J. Virol. 2006, 80, 6738–6744. [Google Scholar] [CrossRef]
- Biris, N.; Tomashevski, A.; Bhattacharya, A.; Diaz-Griffero, F.; Ivanov, D.N. Rhesus monkey TRIM5α SPRY domain recognizes multiple epitopes that span several capsid monomers on the surface of the HIV-1 mature viral core. J. Mol. Biol. 2013, 425, 5032–5044. [Google Scholar] [CrossRef]
- Rahm, N.; Gfeller, D.; Snoeck, J.; Martinez, R.; McLaren, P.J.; Ortiz, M.; Ciuffi, A.; Telenti, A. Susceptibility and adaptation to human TRIM5α alleles at positive selected sites in HIV-1 capsid. Virology 2013, 441, 162–170. [Google Scholar] [CrossRef]
- Roa, A.; Hayashi, F.; Yang, Y.; Lienlaf, M.; Zhou, J.; Shi, J.; Watanabe, S.; Kigawa, T.; Yokoyama, S.; Aiken, C.; et al. RING domain mutations uncouple TRIM5α restriction of HIV-1 from inhibition of reverse transcription and acceleration of uncoating. J. Virol. 2012, 86, 1717–1727. [Google Scholar] [CrossRef]
- Kutluay, S.B.; Perez-Caballero, D.; Bieniasz, P.D. Fates of retroviral core components during unrestricted and TRIM5-restricted infection. PLoS Pathog. 2013, 9, e1003214. [Google Scholar] [CrossRef]
- Anderson, J.L.; Campbell, E.M.; Wu, X.; Vandegraaff, N.; Engelman, A.; Hope, T.J. Proteasome inhibition reveals that a functional preintegration complex intermediate can be generated during restriction by diverse TRIM5 proteins. J. Virol. 2006, 80, 9754–9760. [Google Scholar] [CrossRef]
- Wu, X.; Anderson, J.L.; Campbell, E.M.; Joseph, A.M.; Hope, T.J. Proteasome inhibitors uncouple rhesus TRIM5alpha restriction of HIV-1 reverse transcription and infection. Proc. Natl. Acad. Sci. USA 2006, 103, 7465–7470. [Google Scholar] [CrossRef]
- Asaoka, K.; Ikeda, K.; Hishinuma, T.; Horie-Inoue, K.; Takeda, S.; Inoue, S. A retrovirus restriction factor TRIM5alpha is transcriptionally regulated by interferons. Biochem. Biophys. Res. Commun. 2005, 338, 1950–1956. [Google Scholar] [CrossRef]
- Carthagena, L.; Parise, M.C.; Ringeard, M.; Chelbi-Alix, M.K.; Hazan, U.; Nisole, S. Implication of TRIM alpha and TRIMCyp in interferon-induced anti-retroviral restriction activities. Retrovirology 2008, 5, 59. [Google Scholar] [CrossRef]
- Sakuma, R.; Mael, A.A.; Ikeda, Y. Alpha interferon enhances TRIM5alpha-mediated antiviral activities in human and rhesus monkey cells. J. Virol. 2007, 81, 10201–10206. [Google Scholar] [CrossRef]
- Carthagena, L.; Bergamaschi, A.; Luna, J.M.; David, A.; Uchil, P.D.; Margottin-Goguet, F.; Mothes, W.; Hazan, U.; Transy, C.; Pancino, G.; et al. Human TRIM gene expression in response to interferons. PLoS ONE 2009, 4, e4894. [Google Scholar] [CrossRef]
- Rajsbaum, R.; Stoye, J.P.; O’Garra, A. Type I interferon-dependent and -independent expression of tripartite motif proteins in immune cells. Eur. J. Immunol. 2008, 38, 619–630. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Gabuzda, D.H.; Lawrence, K.; Langhoff, E.; Terwilliger, E.; Dorfman, T.; Haseltine, W.A.; Sodroski, J. Role of vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes. J. Virol. 1992, 66, 6489–6495. [Google Scholar] [Green Version]
- Sheehy, A.M.; Gaddis, N.C.; Malim, M.H. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 2003, 9, 1404–1407. [Google Scholar] [CrossRef]
- Zennou, V.; Perez-Caballero, D.; Göttlinger, H.; Bieniasz, P.D. APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J. Virol. 2004, 78, 12058–12061. [Google Scholar] [CrossRef]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef]
- Lecossier, D.; Bouchonnet, F.; Clavel, F.; Hance, A.J. Hypermutation of HIV-1 DNA in the Absence of the Vif Protein. Science 2003, 300, 1112. [Google Scholar] [CrossRef]
- Suspène, R.; Sommer, P.; Henry, M.; Ferris, S.; Guétard, D.; Pochet, S.; Chester, A.; Navaratnam, N.; Wain-Hobson, S.; Vartanian, J.-P. APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nucleic Acids Res. 2004, 32, 2421–2429. [Google Scholar] [CrossRef] [Green Version]
- Nowarski, R.; Prabhu, P.; Kenig, E.; Smith, Y.; Britan-Rosich, E.; Kotler, M. APOBEC3G inhibits HIV-1 RNA elongation by inactivating the viral trans-activation response element. J. Mol. Biol. 2014, 426, 2840–2853. [Google Scholar] [CrossRef]
- Langlois, M.-A.; Neuberger, M.S. Human APOBEC3G can restrict retroviral infection in avian cells and acts independently of both UNG and SMUG1. J. Virol. 2008, 82, 4660–4664. [Google Scholar] [CrossRef]
- Kaiser, S.M.; Emerman, M. Uracil DNA glycosylase is dispensable for human immunodeficiency virus type 1 replication and does not contribute to the antiviral effects of the cytidine deaminase Apobec3G. J. Virol. 2006, 80, 875–882. [Google Scholar] [CrossRef]
- Yang, B.; Chen, K.; Zhang, C.; Huang, S.; Zhang, H. Virion-associated uracil DNA glycosylase-2 and apurinic/apyrimidinic endonuclease are involved in the degradation of APOBEC3G-edited nascent HIV-1 DNA. J. Biol. Chem. 2007, 282, 11667–11675. [Google Scholar] [CrossRef]
- Pollpeter, D.; Parsons, M.; Sobala, A.E.; Coxhead, S.; Lang, R.D.; Bruns, A.M.; Papaioannou, S.; McDonnell, J.M.; Apolonia, L.; Chowdhury, J.A.; et al. Deep sequencing of HIV-1 reverse transcripts reveals the multifaceted antiviral functions of APOBEC3G. Nat. Microbiol. 2018, 3, 220–233. [Google Scholar] [CrossRef]
- Guo, F.; Cen, S.; Niu, M.; Yang, Y.; Gorelick, R.J.; Kleiman, L. The interaction of APOBEC3G with human immunodeficiency virus type 1 nucleocapsid inhibits tRNA3Lys annealing to viral RNA. J. Virol. 2007, 81, 11322–11331. [Google Scholar] [CrossRef]
- Bishop, K.N.; Verma, M.; Kim, E.-Y.; Wolinsky, S.M.; Malim, M.H. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008, 4, e1000231. [Google Scholar] [CrossRef]
- Chaurasiya, K.R.; McCauley, M.J.; Wang, W.; Qualley, D.F.; Wu, T.; Kitamura, S.; Geertsema, H.; Chan, D.S.B.; Hertz, A.; Iwatani, Y.; et al. Oligomerization transforms human APOBEC3G from an efficient enzyme to a slowly dissociating nucleic acid-binding protein. Nat. Chem. 2014, 6, 28–33. [Google Scholar] [CrossRef]
- Newman, E.N.C.; Holmes, R.K.; Craig, H.M.; Klein, K.C.; Lingappa, J.R.; Malim, M.H.; Sheehy, A.M. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 2005, 15, 166–170. [Google Scholar] [CrossRef]
- Gillick, K.; Pollpeter, D.; Phalora, P.; Kim, E.-Y.; Wolinsky, S.M.; Malim, M.H. Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4(+) T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. J. Virol. 2013, 87, 1508–1517. [Google Scholar] [CrossRef]
- Suspène, R.; Guétard, D.; Henry, M.; Sommer, P.; Wain-Hobson, S.; Vartanian, J.-P. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 8321–8326. [Google Scholar] [CrossRef]
- Mahieux, R.; Suspène, R.; Delebecque, F.; Henry, M.; Schwartz, O.; Wain-Hobson, S.; Vartanian, J.-P. Extensive editing of a small fraction of human T-cell leukemia virus type 1 genomes by four APOBEC3 cytidine deaminases. J. Gen. Virol. 2005, 86, 2489–2494. [Google Scholar] [CrossRef]
- Delebecque, F.; Suspène, R.; Calattini, S.; Casartelli, N.; Saïb, A.; Froment, A.; Wain-Hobson, S.; Gessain, A.; Vartanian, J.-P.; Schwartz, O. Restriction of foamy viruses by APOBEC cytidine deaminases. J. Virol. 2006, 80, 605–614. [Google Scholar] [CrossRef]
- Browne, E.P.; Littman, D.R. Species-specific restriction of apobec3-mediated hypermutation. J. Virol. 2008, 82, 1305–1313. [Google Scholar] [CrossRef]
- Okeoma, C.M.; Lovsin, N.; Peterlin, B.M.; Ross, S.R. APOBEC3 inhibits mouse mammary tumour virus replication in vivo. Nature 2007, 445, 927–930. [Google Scholar] [CrossRef]
- Koning, F.A.; Newman, E.N.C.; Kim, E.-Y.; Kunstman, K.J.; Wolinsky, S.M.; Malim, M.H. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 2009, 83, 9474–9485. [Google Scholar] [CrossRef]
- Oliva, H.; Pacheco, R.; Martinez-Navio, J.M.; Rodríguez-García, M.; Naranjo-Gómez, M.; Climent, N.; Prado, C.; Gil, C.; Plana, M.; García, F.; et al. Increased expression with differential subcellular location of cytidine deaminase APOBEC3G in human CD4(+) T-cell activation and dendritic cell maturation. Immunol. Cell Biol. 2016, 94, 689–700. [Google Scholar] [CrossRef]
- Peng, G.; Lei, K.J.; Jin, W.; Greenwell-Wild, T.; Wahl, S.M. Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J. Exp. Med. 2006, 203, 41–46. [Google Scholar] [CrossRef]
- Peng, G.; Greenwell-Wild, T.; Nares, S.; Jin, W.; Lei, K.J.; Rangel, Z.G.; Munson, P.J.; Wahl, S.M. Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood 2007, 110, 393–400. [Google Scholar] [CrossRef]
- Sarkis, P.T.N.; Ying, S.; Xu, R.; Yu, X.-F. STAT1-independent cell type-specific regulation of antiviral APOBEC3G by IFN-alpha. J. Immunol. 2006, 177, 4530–4540. [Google Scholar] [CrossRef]
- Chen, K.; Huang, J.; Zhang, C.; Huang, S.; Nunnari, G.; Wang, F.; Tong, X.; Gao, L.; Nikisher, K.; Zhang, H. Alpha interferon potently enhances the anti-human immunodeficiency virus type 1 activity of APOBEC3G in resting primary CD4 T cells. J. Virol. 2006, 80, 7645–7657. [Google Scholar] [CrossRef]
- Pillai, S.K.; Abdel-Mohsen, M.; Guatelli, J.; Skasko, M.; Monto, A.; Fujimoto, K.; Yukl, S.; Greene, W.C.; Kovari, H.; Rauch, A.; et al. Role of retroviral restriction factors in the interferon-α-mediated suppression of HIV-1 in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 3035–3040. [Google Scholar] [CrossRef]
- Wang, F.-X.; Huang, J.; Zhang, H.; Ma, X.; Zhang, H. APOBEC3G upregulation by alpha interferon restricts human immunodeficiency virus type 1 infection in human peripheral plasmacytoid dendritic cells. J. Gen. Virol. 2008, 89, 722–730. [Google Scholar] [CrossRef]
- Mohanram, V.; Sköld, A.E.; Bächle, S.M.; Pathak, S.K.; Spetz, A.-L. IFN-α induces APOBEC3G, F, and A in immature dendritic cells and limits HIV-1 spread to CD4+ T cells. J. Immunol. 2013, 190, 3346–3353. [Google Scholar] [CrossRef]
- Argyris, E.G.; Acheampong, E.; Wang, F.; Huang, J.; Chen, K.; Mukhtar, M.; Zhang, H. The interferon-induced expression of APOBEC3G in human blood-brain barrier exerts a potent intrinsic immunity to block HIV-1 entry to central nervous system. Virology 2007, 367, 440–451. [Google Scholar] [CrossRef]
- Trapp, S.; Derby, N.R.; Singer, R.; Shaw, A.; Williams, V.G.; Turville, S.G.; Bess, J.W.; Lifson, J.D.; Robbiani, M. Double-stranded RNA analog poly(I:C) inhibits human immunodeficiency virus amplification in dendritic cells via type I interferon-mediated activation of APOBEC3G. J. Virol. 2009, 83, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, X.; Liu, M.; Hu, Q.; Song, L.; Ye, L.; Zhou, D.; Ho, W. A critical function of toll-like receptor-3 in the induction of anti-human immunodeficiency virus activities in macrophages. Immunology 2010, 131, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.O.; Suzuki, T.; Yamamoto, N.; Watanabe, M.; Takaku, H. HIV-1 Gag-virus-like particles inhibit HIV-1 replication in dendritic cells and T cells through IFN-α-dependent upregulation of APOBEC3G and 3F. J. Innate. Immun. 2012, 4, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Stopak, K.S.; Chiu, Y.-L.; Kropp, J.; Grant, R.M.; Greene, W.C. Distinct patterns of cytokine regulation of APOBEC3G expression and activity in primary lymphocytes, macrophages, and dendritic cells. J. Biol. Chem. 2007, 282, 3539–3546. [Google Scholar] [CrossRef] [PubMed]
- Goldstone, D.C.; Ennis-Adeniran, V.; Hedden, J.J.; Groom, H.C.T.; Rice, G.I.; Christodoulou, E.; Walker, P.A.; Kelly, G.; Haire, L.F.; Yap, M.W.; et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011, 480, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Powell, R.D.; Holland, P.J.; Hollis, T.; Perrino, F.W. Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J. Biol. Chem. 2011, 286, 43596–43600. [Google Scholar] [CrossRef] [PubMed]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Baldauf, H.-M.; Pan, X.; Erikson, E.; Schmidt, S.; Daddacha, W.; Burggraf, M.; Schenkova, K.; Ambiel, I.; Wabnitz, G.; Gramberg, T.; et al. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat. Med. 2012, 18, 1682–1687. [Google Scholar] [CrossRef]
- Descours, B.; Cribier, A.; Chable-Bessia, C.; Ayinde, D.; Rice, G.; Crow, Y.; Yatim, A.; Schwartz, O.; Laguette, N.; Benkirane, M. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4(+) T-cells. Retrovirology 2012, 9, 87. [Google Scholar] [CrossRef]
- Valle-Casuso, J.C.; Allouch, A.; David, A.; Lenzi, G.M.; Studdard, L.; Barré-Sinoussi, F.; Müller-Trutwin, M.; Kim, B.; Pancino, G.; Sáez-Cirión, A. p21 restricts HIV-1 in monocyte-derived dendritic cells through the reduction of deoxynucleoside triphosphate biosynthesis and regulation of SAMHD1 antiviral activity. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Jáuregui, P.; Landau, N.R. DNA damage induces a SAMHD1-mediated block to the infection of macrophages by HIV-1. Sci. Rep. 2018, 8, 4153. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, S.; Daly, M.B.; St Gelais, C.; Kim, S.H.; Hollenbaugh, J.A.; Shepard, C.; Kennedy, E.M.; Kim, D.-H.; Schinazi, R.F.; Kim, B.; et al. SAMHD1 controls cell cycle status, apoptosis and HIV-1 infection in monocytic THP-1 cells. Virology 2016, 495, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Pauls, E.; Ruiz, A.; Badia, R.; Permanyer, M.; Gubern, A.; Riveira-Muñoz, E.; Torres-Torronteras, J.; Alvarez, M.; Mothe, B.; Brander, C.; et al. Cell cycle control and HIV-1 susceptibility are linked by CDK6-dependent CDK2 phosphorylation of SAMHD1 in myeloid and lymphoid cells. J. Immunol. 2014, 193, 1988–1997. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Hao, C.; DeLucia, M.; Swanson, S.; Florens, L.; Washburn, M.P.; Ahn, J.; Skowronski, J. CyclinA2-cyclin-dependent kinase regulates SAMHD1 protein phosphohydrolase domain. J. Biol. Chem. 2015, 290, 13279–13292. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Pauls, E.; Badia, R.; Torres-Torronteras, J.; Riveira-Muñoz, E.; Clotet, B.; Martí, R.; Ballana, E.; Esté, J.A. Cyclin D3-dependent control of the dNTP pool and HIV-1 replication in human macrophages. Cell Cycle 2015, 14, 1657–1665. [Google Scholar] [CrossRef]
- White, T.E.; Brandariz-Nuñez, A.; Valle-Casuso, J.C.; Amie, S.; Nguyen, L.A.; Kim, B.; Tuzova, M.; Diaz-Griffero, F. The retroviral restriction ability of SAMHD1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation. Cell Host Microbe 2013, 13, 441–451. [Google Scholar] [CrossRef]
- Arnold, L.H.; Groom, H.C.T.; Kunzelmann, S.; Schwefel, D.; Caswell, S.J.; Ordonez, P.; Mann, M.C.; Rueschenbaum, S.; Goldstone, D.C.; Pennell, S.; et al. Phospho-dependent Regulation of SAMHD1 Oligomerisation Couples Catalysis and Restriction. PLoS Pathog. 2015, 11, e1005194. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Wang, Z.; White, T.; Buffone, C.; Nguyen, L.A.; Shepard, C.N.; Kim, B.; Demeler, B.; Diaz-Griffero, F.; Ivanov, D.N. Effects of T592 phosphomimetic mutations on tetramer stability and dNTPase activity of SAMHD1 can not explain the retroviral restriction defect. Sci. Rep. 2016, 6, 31353. [Google Scholar] [CrossRef] [Green Version]
- Majer, C.; Schüssler, J.M.; König, R. Intertwined: SAMHD1 cellular functions, restriction, and viral evasion strategies. Med. Microbiol. Immunol. 2019. [Google Scholar] [CrossRef]
- Cribier, A.; Descours, B.; Valadão, A.L.C.; Laguette, N.; Benkirane, M. Phosphorylation of SAMHD1 by cyclin A2/CDK1 regulates its restriction activity toward HIV-1. Cell Rep. 2013, 3, 1036–1043. [Google Scholar] [CrossRef]
- Coiras, M.; Bermejo, M.; Descours, B.; Mateos, E.; García-Pérez, J.; López-Huertas, M.-R.; Lederman, M.M.; Benkirane, M.; Alcamí, J. IL-7 induces SAMHD1 phosphorylation in CD4+ T lymphocytes, improving early steps of HIV-1 life cycle. Cell Rep. 2016, 14, 2100–2107. [Google Scholar] [CrossRef] [PubMed]
- Beloglazova, N.; Flick, R.; Tchigvintsev, A.; Brown, G.; Popovic, A.; Nocek, B.; Yakunin, A.F. Nuclease activity of the human SAMHD1 protein implicated in the Aicardi-Goutieres syndrome and HIV-1 restriction. J. Biol. Chem. 2013, 288, 8101–8110. [Google Scholar] [CrossRef] [PubMed]
- Seamon, K.J.; Sun, Z.; Shlyakhtenko, L.S.; Lyubchenko, Y.L.; Stivers, J.T. SAMHD1 is a single-stranded nucleic acid binding protein with no active site-associated nuclease activity. Nucleic Acids Res. 2015, 43, 6486–6499. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, J.; Choi, J.; Oh, C.; Kim, S.; Seo, M.; Kim, S.-Y.; Seo, D.; Kim, J.; White, T.E.; Brandariz-Nuñez, A.; et al. The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat. Med. 2014, 20, 936–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Ryoo, J.; Oh, C.; Hwang, S.; Ahn, K. SAMHD1 specifically restricts retroviruses through its RNase activity. Retrovirology 2015, 12, 46. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, J.M.; St Gelais, C.; de Silva, S.; Yount, J.S.; Tang, C.; Ji, X.; Shepard, C.; Xiong, Y.; Kim, B.; Wu, L. SAMHD1-mediated HIV-1 restriction in cells does not involve ribonuclease activity. Nat. Med. 2016, 22, 1072–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, T.E.; Brandariz-Nuñez, A.; Valle-Casuso, J.C.; Amie, S.; Nguyen, L.; Kim, B.; Brojatsch, J.; Diaz-Griffero, F. Contribution of SAM and HD domains to retroviral restriction mediated by human SAMHD1. Virology 2013, 436, 81–90. [Google Scholar] [CrossRef]
- Gramberg, T.; Kahle, T.; Bloch, N.; Wittmann, S.; Müllers, E.; Daddacha, W.; Hofmann, H.; Kim, B.; Lindemann, D.; Landau, N.R. Restriction of diverse retroviruses by SAMHD1. Retrovirology 2013, 10, 26. [Google Scholar] [CrossRef]
- Sze, A.; Belgnaoui, S.M.; Olagnier, D.; Lin, R.; Hiscott, J.; van Grevenynghe, J. Host restriction factor SAMHD1 limits human T cell leukemia virus type 1 infection of monocytes via STING-mediated apoptosis. Cell Host Microbe 2013, 14, 422–434. [Google Scholar] [CrossRef]
- Hollenbaugh, J.A.; Gee, P.; Baker, J.; Daly, M.B.; Amie, S.M.; Tate, J.; Kasai, N.; Kanemura, Y.; Kim, D.-H.; Ward, B.M.; et al. Host factor SAMHD1 restricts DNA viruses in non-dividing myeloid cells. PLoS Pathog. 2013, 9, e1003481. [Google Scholar] [CrossRef]
- St Gelais, C.; de Silva, S.; Amie, S.M.; Coleman, C.M.; Hoy, H.; Hollenbaugh, J.A.; Kim, B.; Wu, L. SAMHD1 restricts HIV-1 infection in dendritic cells (DCs) by dNTP depletion, but its expression in DCs and primary CD4+ T-lymphocytes cannot be upregulated by interferons. Retrovirology 2012, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhan, Y.; Zhou, Y.; Jiang, Y.; Zheng, X.; Yu, L.; Tong, W.; Gao, F.; Li, L.; Huang, Q.; et al. Interferon regulatory factor 3 is a key regulation factor for inducing the expression of SAMHD1 in antiviral innate immunity. Sci. Rep. 2016, 6, 29665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Zhu, M.; Pan, X.; Zhu, Y.; Yan, H.; Jiang, T.; Shen, Y.; Dong, X.; Zheng, N.; Lu, J.; et al. Inhibition of Hepatitis B virus replication by SAMHD1. Biochem. Biophys. Res. Commun. 2014, 450, 1462–1468. [Google Scholar] [CrossRef]
- Zhu, M.; Lu, J.; Dong, X.; Zheng, N.; Li, T.; Chen, Z.; Pan, X.; Zhu, Y.; Yan, H.; Shen, Y.; et al. Interferon-stimulated gene factor 3 complex is required for the induction of sterile α motif and HD domain-containing protein 1 expression by interferon-α in SMMC-7721 cells. Mol. Med. Rep. 2015, 12, 7176–7180. [Google Scholar] [CrossRef] [PubMed]
- Pauls, E.; Jimenez, E.; Ruiz, A.; Permanyer, M.; Ballana, E.; Costa, H.; Nascimiento, R.; Parkhouse, R.M.; Peña, R.; Riveiro-Muñoz, E.; et al. Restriction of HIV-1 replication in primary macrophages by IL-12 and IL-18 through the upregulation of SAMHD1. J. Immunol. 2013, 190, 4736–4741. [Google Scholar] [CrossRef]
- Rennie, M.L.; McKelvie, S.A.; Bulloch, E.M.M.; Kingston, R.L. Transient dimerization of human MxA promotes GTP hydrolysis, resulting in a mechanical power stroke. Structure 2014, 22, 1433–1445. [Google Scholar] [CrossRef]
- Aebi, M.; Fäh, J.; Hurt, N.; Samuel, C.E.; Thomis, D.; Bazzigher, L.; Pavlovic, J.; Haller, O.; Staeheli, P. cDNA structures and regulation of two interferon-induced human Mx proteins. Mol. Cell. Biol. 1989, 9, 5062–5072. [Google Scholar] [CrossRef]
- Horisberger, M.A.; McMaster, G.K.; Zeller, H.; Wathelet, M.G.; Dellis, J.; Content, J. Cloning and sequence analyses of cDNAs for interferon- and virus-induced human Mx proteins reveal that they contain putative guanine nucleotide-binding sites: Functional study of the corresponding gene promoter. J. Virol. 1990, 64, 1171–1181. [Google Scholar]
- al-Masri, A.N.; Werfel, T.; Jakschies, D.; von Wussow, P. Intracellular staining of Mx proteins in cells from peripheral blood, bone marrow and skin. MP Mol. Pathol. 1997, 50, 9–14. [Google Scholar] [CrossRef]
- Sanda, C.; Weitzel, P.; Tsukahara, T.; Schaley, J.; Edenberg, H.J.; Stephens, M.A.; McClintick, J.N.; Blatt, L.M.; Li, L.; Brodsky, L.; et al. Differential gene induction by type I and type II interferons and their combination. J. Interferon Cytokine Res. 2006, 26, 462–472. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Liu, M.-Q.; Li, J.-L.; Zhou, R.-H.; Zhou, Y.; Wang, Y.-Z.; Zhou, W.; Ho, W.-Z. IFN-λ inhibits drug-resistant HIV infection of macrophages. Front. Immunol. 2017, 8, 210. [Google Scholar] [CrossRef]
- Kane, M.; Yadav, S.S.; Bitzegeio, J.; Kutluay, S.B.; Zang, T.; Wilson, S.J.; Schoggins, J.W.; Rice, C.M.; Yamashita, M.; Hatziioannou, T.; et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 2013, 502, 563–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matreyek, K.A.; Wang, W.; Serrao, E.; Singh, P.K.; Levin, H.L.; Engelman, A. Host and viral determinants for MxB restriction of HIV-1 infection. Retrovirology 2014, 11, 90. [Google Scholar] [CrossRef] [PubMed]
- Fricke, T.; White, T.E.; Schulte, B.; de Souza Aranha Vieira, D.A.; Dharan, A.; Campbell, E.M.; Brandariz-Nuñez, A.; Diaz-Griffero, F. MxB binds to the HIV-1 core and prevents the uncoating process of HIV-1. Retrovirology 2014, 11, 68. [Google Scholar] [CrossRef] [PubMed]
- Schulte, B.; Buffone, C.; Opp, S.; Di Nunzio, F.; De Souza Aranha Vieira, D.A.; Brandariz-Nuñez, A.; Diaz-Griffero, F. Restriction of HIV-1 requires the N-terminal region of MxB as a capsid-binding motif but not as a nuclear localization signal. J. Virol. 2015, 89, 8599–8610. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Pan, Q.; Ding, S.; Qian, J.; Xu, F.; Zhou, J.; Cen, S.; Guo, F.; Liang, C. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 2013, 14, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Dicks, M.D.J.; Goujon, C.; Pollpeter, D.; Betancor, G.; Apolonia, L.; Bergeron, J.R.C.; Malim, M.H. Oligomerization requirements for MX2-mediated suppression of HIV-1 infection. J. Virol. 2016, 90, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Buffone, C.; Schulte, B.; Opp, S.; Diaz-Griffero, F. Contribution of MxB oligomerization to HIV-1 capsid binding and restriction. J. Virol. 2015, 89, 3285–3294. [Google Scholar] [CrossRef]
- Alvarez, F.J.D.; He, S.; Perilla, J.R.; Jang, S.; Schulten, K.; Engelman, A.N.; Scheres, S.H.W.; Zhang, P. CryoEM structure of MxB reveals a novel oligomerization interface critical for HIV restriction. Sci. Adv. 2017, 3, e1701264. [Google Scholar] [CrossRef]
- Buffone, C.; Kutzner, J.; Opp, S.; Martinez-Lopez, A.; Selyutina, A.; Coggings, S.A.; Studdard, L.R.; Ding, L.; Kim, B.; Spearman, P.; et al. The ability of SAMHD1 to block HIV-1 but not SIV requires expression of MxB. Virology 2019, 531, 260–268. [Google Scholar] [CrossRef]
- Sironi, M.; Biasin, M.; Cagliani, R.; Gnudi, F.; Saulle, I.; Ibba, S.; Filippi, G.; Yahyaei, S.; Tresoldi, C.; Riva, S.; et al. Evolutionary analysis identifies an MX2 haplotype associated with natural resistance to HIV-1 infection. Mol. Biol. Evol. 2014, 31, 2402–2414. [Google Scholar] [CrossRef] [PubMed]
- Crameri, M.; Bauer, M.; Caduff, N.; Walker, R.; Steiner, F.; Franzoso, F.D.; Gujer, C.; Boucke, K.; Kucera, T.; Zbinden, A.; et al. MxB is an interferon-induced restriction factor of human herpesviruses. Nat. Commun. 2018, 9, 1980. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Luo, F.; Chen, Q.; Zhu, N.; Wang, H.; Xie, L.; Xiong, H.; Yue, M.; Zhang, Y.; Feng, Y.; et al. IFN-λs inhibit Hantaan virus infection through the JAK-STAT pathway and expression of Mx2 protein. Genes Immun. 2019, 20, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Kupzig, S.; Korolchuk, V.; Rollason, R.; Sugden, A.; Wilde, A.; Banting, G. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic 2003, 4, 694–709. [Google Scholar] [CrossRef] [PubMed]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, K.; Skasko, M.; Deerinck, T.J.; Crum, J.; Ellisman, M.H.; Guatelli, J. Direct restriction of virus release and incorporation of the interferon-induced protein BST-2 into HIV-1 particles. PLoS Pathog. 2010, 6, e1000701. [Google Scholar] [CrossRef] [PubMed]
- Hammonds, J.; Wang, J.-J.; Yi, H.; Spearman, P. Immunoelectron microscopic evidence for Tetherin/BST2 as the physical bridge between HIV-1 virions and the plasma membrane. PLoS Pathog. 2010, 6, e1000749. [Google Scholar] [CrossRef]
- Venkatesh, S.; Bieniasz, P.D. Mechanism of HIV-1 virion entrapment by tetherin. PLoS Pathog. 2013, 9, e1003483. [Google Scholar] [CrossRef]
- Miyakawa, K.; Ryo, A.; Murakami, T.; Ohba, K.; Yamaoka, S.; Fukuda, M.; Guatelli, J.; Yamamoto, N. BCA2/Rabring7 promotes tetherin-dependent HIV-1 restriction. PLoS Pathog. 2009, 5, e1000700. [Google Scholar] [CrossRef]
- Cocka, L.J.; Bates, P. Identification of alternatively translated Tetherin isoforms with differing antiviral and signaling activities. PLoS Pathog. 2012, 8, e1002931. [Google Scholar] [CrossRef]
- Erikson, E.; Adam, T.; Schmidt, S.; Lehmann-Koch, J.; Over, B.; Goffinet, C.; Harter, C.; Bekeredjian-Ding, I.; Sertel, S.; Lasitschka, F.; et al. In vivo expression profile of the antiviral restriction factor and tumor-targeting antigen CD317/BST-2/HM1.24/tetherin in humans. Proc. Natl. Acad. Sci. USA 2011, 108, 13688–13693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtomo, T.; Sugamata, Y.; Ozaki, Y.; Ono, K.; Yoshimura, Y.; Kawai, S.; Koishihara, Y.; Ozaki, S.; Kosaka, M.; Hirano, T.; et al. Molecular cloning and characterization of a surface antigen preferentially overexpressed on multiple myeloma cells. Biochem. Biophys. Res. Commun. 1999, 258, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Park, S.-H.; Ye, S.-K.; Kim, M. IFN-γ-induced BST2 mediates monocyte adhesion to human endothelial cells. Cell. Immunol. 2011, 267, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Amet, T.; Byrd, D.; Hu, N.; Sun, Q.; Li, F.; Zhao, Y.; Hu, S.; Grantham, A.; Yu, Q. BST-2 expression in human hepatocytes is inducible by all three types of interferons and restricts production of hepatitis C virus. Curr. Mol. Med. 2014, 14, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A.M.; Miller, K.D.; Cavanaugh, S.E.; Rall, G.F. Bst2/Tetherin is induced in neurons by type I interferon and viral infection but is dispensable for protection against neurotropic viral challenge. J. Virol. 2015, 89, 11011–11018. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, F.P.; Stalder, R.; Czubala, M.; Lehmann, M.; Rio, L.; Mangeat, B.; Piguet, V. TLR-4 engagement of dendritic cells confers a BST-2/tetherin-mediated restriction of HIV-1 infection to CD4+ T cells across the virological synapse. Retrovirology 2013, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Homann, S.; Smith, D.; Little, S.; Richman, D.; Guatelli, J. Upregulation of BST-2/Tetherin by HIV infection in vivo. J. Virol. 2011, 85, 10659–10668. [Google Scholar] [CrossRef] [PubMed]
- Guzzo, C.; Jung, M.; Graveline, A.; Banfield, B.W.; Gee, K. IL-27 increases BST-2 expression in human monocytes and T cells independently of type I IFN. Sci. Rep. 2012, 2, 974. [Google Scholar] [CrossRef]
- Bego, M.G.; Mercier, J.; Cohen, E.A. Virus-activated interferon regulatory factor 7 upregulates expression of the interferon-regulated BST2 gene independently of interferon signaling. J. Virol. 2012, 86, 3513–3527. [Google Scholar] [CrossRef]
- Bauman, D.R.; Bitmansour, A.D.; McDonald, J.G.; Thompson, B.M.; Liang, G.; Russell, D.W. 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc. Natl. Acad. Sci. USA 2009, 106, 16764–16769. [Google Scholar] [CrossRef] [Green Version]
- Park, K.; Scott, A.L. Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J. Leukoc. Biol. 2010, 88, 1081–1087. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.-Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 2013, 38, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, S.; Yi, Z.; Tian, H.; Aliyari, R.; Li, Y.; Chen, G.; Liu, P.; Zhong, J.; Chen, X.; et al. Interferon-inducible cholesterol-25-hydroxylase inhibits hepatitis C virus replication via distinct mechanisms. Sci. Rep. 2014, 4, 7242. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Deng, Y.-Q.; Wang, S.; Ma, F.; Aliyari, R.; Huang, X.-Y.; Zhang, N.-N.; Watanabe, M.; Dong, H.-L.; Liu, P.; et al. 25-Hydroxycholesterol protects host against zika virus infection and its associated microcephaly in a mouse model. Immunity 2017, 46, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Gomes, B.; Gonçalves, S.; Disalvo, A.; Hollmann, A.; Santos, N.C. Effect of 25-hydroxycholesterol in viral membrane fusion: Insights on HIV inhibition. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Guo, X.; Goff, S.P. Inhibition of retroviral RNA production by ZAP, a CCCH-type zinc finger protein. Science 2002, 297, 1703–1706. [Google Scholar] [CrossRef]
- Bick, M.J.; Carroll, J.-W.N.; Gao, G.; Goff, S.P.; Rice, C.M.; MacDonald, M.R. Expression of the zinc-finger antiviral protein inhibits alphavirus replication. J. Virol. 2003, 77, 11555–11562. [Google Scholar] [CrossRef]
- Müller, S.; Möller, P.; Bick, M.J.; Wurr, S.; Becker, S.; Günther, S.; Kümmerer, B.M. Inhibition of filovirus replication by the zinc finger antiviral protein. J. Virol. 2007, 81, 2391–2400. [Google Scholar] [CrossRef]
- Mao, R.; Nie, H.; Cai, D.; Zhang, J.; Liu, H.; Yan, R.; Cuconati, A.; Block, T.M.; Guo, J.-T.; Guo, H. Inhibition of hepatitis B virus replication by the host zinc finger antiviral protein. PLoS Pathog. 2013, 9, e1003494. [Google Scholar] [CrossRef]
- Li, M.; Yan, K.; Wei, L.; Yang, J.; Lu, C.; Xiong, F.; Zheng, C.; Xu, W. Zinc finger antiviral protein inhibits coxsackievirus B3 virus replication and protects against viral myocarditis. Antiviral Res. 2015, 123, 50–61. [Google Scholar] [CrossRef]
- Chiu, H.-P.; Chiu, H.; Yang, C.-F.; Lee, Y.-L.; Chiu, F.-L.; Kuo, H.-C.; Lin, R.-J.; Lin, Y.-L. Inhibition of Japanese encephalitis virus infection by the host zinc-finger antiviral protein. PLoS Pathog. 2018, 14, e1007166. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Carroll, J.-W.N.; Macdonald, M.R.; Goff, S.P.; Gao, G. The zinc finger antiviral protein directly binds to specific viral mRNAs through the CCCH zinc finger motifs. J. Virol. 2004, 78, 12781–12787. [Google Scholar] [CrossRef]
- Guo, X.; Ma, J.; Sun, J.; Gao, G. The zinc-finger antiviral protein recruits the RNA processing exosome to degrade the target mRNA. Proc. Natl. Acad. Sci. USA 2007, 104, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Guo, X.; Lv, F.; Xu, Y.; Gao, G. p72 DEAD box RNA helicase is required for optimal function of the zinc-finger antiviral protein. Proc. Natl. Acad. Sci. USA 2008, 105, 4352–4357. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Chen, G.; Lv, F.; Wang, X.; Ji, X.; Xu, Y.; Sun, J.; Wu, L.; Zheng, Y.-T.; Gao, G. Zinc-finger antiviral protein inhibits HIV-1 infection by selectively targeting multiply spliced viral mRNAs for degradation. Proc. Natl. Acad. Sci. USA 2011, 108, 15834–15839. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Wang, X.; Goff, S.P.; Gao, G. Translational repression precedes and is required for ZAP-mediated mRNA decay. EMBO J. 2012, 31, 4236–4246. [Google Scholar] [CrossRef]
- Zheng, X.; Wang, X.; Tu, F.; Wang, Q.; Fan, Z.; Gao, G. TRIM25 is required for the antiviral activity of zinc finger antiviral protein. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.H.; Lau, Z.; Cheung, P.; Aguilar, E.G.; Schneider, W.M.; Bozzacco, L.; Molina, H.; Buehler, E.; Takaoka, A.; Rice, C.M.; et al. TRIM25 enhances the antiviral action of zinc-finger antiviral protein (ZAP). PLoS Pathog. 2017, 13, e1006145. [Google Scholar] [CrossRef]
- Kerns, J.A.; Emerman, M.; Malik, H.S. Positive selection and increased antiviral activity associated with the PARP-containing isoform of human zinc-finger antiviral protein. PLoS Genet. 2008, 4, e21. [Google Scholar] [CrossRef]
- Hayakawa, S.; Shiratori, S.; Yamato, H.; Kameyama, T.; Kitatsuji, C.; Kashigi, F.; Goto, S.; Kameoka, S.; Fujikura, D.; Yamada, T.; et al. ZAPS is a potent stimulator of signaling mediated by the RNA helicase RIG-I during antiviral responses. Nat. Immunol. 2011, 12, 37–44. [Google Scholar] [CrossRef]
- Li, M.M.H.; Aguilar, E.G.; Michailidis, E.; Pabon, J.; Park, P.; Wu, X.; de Jong, Y.P.; Schneider, W.M.; Molina, H.; Rice, C.M.; et al. Characterization of novel splice variants of zinc finger antiviral protein (ZAP). J. Virol. 2019. [Google Scholar] [CrossRef]
- Erazo, A.; Goff, S.P. Nuclear matrix protein Matrin 3 is a regulator of ZAP-mediated retroviral restriction. Retrovirology 2015, 12, 57. [Google Scholar] [CrossRef]
- Yedavalli, V.S.R.K.; Jeang, K.-T. Matrin 3 is a co-factor for HIV-1 Rev in regulating post-transcriptional viral gene expression. Retrovirology 2011, 8, 61. [Google Scholar] [CrossRef]
- van Weringh, A.; Ragonnet-Cronin, M.; Pranckeviciene, E.; Pavon-Eternod, M.; Kleiman, L.; Xia, X. HIV-1 modulates the tRNA pool to improve translation efficiency. Mol. Biol. Evol. 2011, 28, 1827–1834. [Google Scholar] [CrossRef]
- Lin, Y.-Z.; Sun, L.-K.; Zhu, D.-T.; Hu, Z.; Wang, X.-F.; Du, C.; Wang, Y.-H.; Wang, X.-J.; Zhou, J.-H. Equine schlafen 11 restricts the production of equine infectious anemia virus via a codon usage-dependent mechanism. Virology 2016, 495, 112–121. [Google Scholar] [CrossRef]
- Stabell, A.C.; Hawkins, J.; Li, M.; Gao, X.; David, M.; Press, W.H.; Sawyer, S.L. Non-human primate Schlafen11 inhibits production of both host and viral proteins. PLoS Pathog. 2016, 12, e1006066. [Google Scholar] [CrossRef]
- Li, J.; Peet, G.W.; Balzarano, D.; Li, X.; Massa, P.; Barton, R.W.; Marcu, K.B. Novel NEMO/IκB kinase and NF-κB target genes at the Pre-B to immature B cell transition. J. Biol. Chem. 2001, 276, 18579–18590. [Google Scholar] [CrossRef]
- Sen, G.C.; Sarkar, S.N. The interferon-stimulated genes: Targets of direct signaling by interferons, double-stranded RNA, and viruses. In Interferon: The 50th Anniversary; Pitha, P.M., Ed.; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2007; pp. 233–250. ISBN 978-3-540-71329-6. [Google Scholar]
- Villarroya-Beltri, C.; Guerra, S.; Sánchez-Madrid, F. ISGylation—A key to lock the cell gates for preventing the spread of threats. J. Cell. Sci. 2017, 130, 2961–2969. [Google Scholar] [CrossRef]
- Okumura, A.; Lu, G.; Pitha-Rowe, I.; Pitha, P.M. Innate antiviral response targets HIV-1 release by the induction of ubiquitin-like protein ISG15. Proc. Natl. Acad. Sci. USA 2006, 103, 1440–1445. [Google Scholar] [CrossRef] [Green Version]
- Malakhova, O.A.; Zhang, D.-E. ISG15 inhibits Nedd4 ubiquitin E3 activity and enhances the innate antiviral response. J. Biol. Chem. 2008, 283, 8783–8787. [Google Scholar] [CrossRef]
- Wong, J.J.Y.; Pung, Y.F.; Sze, N.S.-K.; Chin, K.-C. HERC5 is an IFN-induced HECT-type E3 protein ligase that mediates type I IFN-induced ISGylation of protein targets. Proc. Natl. Acad. Sci. USA 2006, 103, 10735–10740. [Google Scholar] [CrossRef] [Green Version]
- Woods, M.W.; Kelly, J.N.; Hattlmann, C.J.; Tong, J.G.K.; Xu, L.S.; Coleman, M.D.; Quest, G.R.; Smiley, J.R.; Barr, S.D. Human HERC5 restricts an early stage of HIV-1 assembly by a mechanism correlating with the ISGylation of Gag. Retrovirology 2011, 8, 95. [Google Scholar] [CrossRef]
- Pincetic, A.; Kuang, Z.; Seo, E.J.; Leis, J. The interferon-induced gene ISG15 blocks retrovirus release from cells late in the budding process. J. Virol. 2010, 84, 4725–4736. [Google Scholar] [CrossRef]
- Scagnolari, C.; Monteleone, K.; Selvaggi, C.; Pierangeli, A.; D’Ettorre, G.; Mezzaroma, I.; Turriziani, O.; Gentile, M.; Vullo, V.; Antonelli, G. ISG15 expression correlates with HIV-1 viral load and with factors regulating T cell response. Immunobiology 2016, 221, 282–290. [Google Scholar] [CrossRef]
- Gargan, S.; Ahmed, S.; Mahony, R.; Bannan, C.; Napoletano, S.; O’Farrelly, C.; Borrow, P.; Bergin, C.; Stevenson, N.J. HIV-1 promotes the degradation of components of the type 1 IFN JAK/STAT pathway and blocks anti-viral ISG induction. EBioMedicine 2018, 30, 203–216. [Google Scholar] [CrossRef]
- Vestal, D.J.; Jeyaratnam, J.A. The guanylate-binding proteins: Emerging insights into the biochemical properties and functions of this family of large interferon-induced guanosine triphosphatase. J. Interferon Cytokine Res. 2011, 31, 89–97. [Google Scholar] [CrossRef]
- McLaren, P.J.; Gawanbacht, A.; Pyndiah, N.; Krapp, C.; Hotter, D.; Kluge, S.F.; Götz, N.; Heilmann, J.; Mack, K.; Sauter, D.; et al. Identification of potential HIV restriction factors by combining evolutionary genomic signatures with functional analyses. Retrovirology 2015, 12, 41. [Google Scholar] [CrossRef]
- Krapp, C.; Hotter, D.; Gawanbacht, A.; McLaren, P.J.; Kluge, S.F.; Stürzel, C.M.; Mack, K.; Reith, E.; Engelhart, S.; Ciuffi, A.; et al. Guanylate binding protein (GBP) 5 is an interferon-inducible inhibitor of HIV-1 infectivity. Cell Host Microbe 2016, 19, 504–514. [Google Scholar] [CrossRef]
- Hotter, D.; Sauter, D.; Kirchhoff, F. Guanylate binding protein 5: Impairing virion infectivity by targeting retroviral envelope glycoproteins. Small GTPases 2017, 8, 31–37. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Hizukuri, Y.; Yamashiro, K.; Makita, N.; Ohnishi, K.; Takeya, M.; Komohara, Y.; Hayashi, Y. Guanylate-binding protein 5 is a marker of interferon-γ-induced classically activated macrophages. Clin. Transl. Immunol. 2016, 5, e111. [Google Scholar] [CrossRef]
- Richards, K.H.; Clapham, P.R. Effects of vpu start-codon mutations on human immunodeficiency virus type 1 replication in macrophages. J. Gen. Virol. 2007, 88, 2780–2792. [Google Scholar] [CrossRef]
- Alteber, Z.; Sharbi-Yunger, A.; Pevsner-Fischer, M.; Blat, D.; Roitman, L.; Tzehoval, E.; Elinav, E.; Eisenbach, L. The anti-inflammatory IFITM genes ameliorate colitis and partially protect from tumorigenesis by changing immunity and microbiota. Immunol. Cell Biol. 2018, 96, 284–297. [Google Scholar] [CrossRef]
- Yánez, D.C.; Sahni, H.; Ross, S.; Solanki, A.; Lau, C.-I.; Papaioannou, E.; Barbarulo, A.; Powell, R.; Lange, U.C.; Adams, D.J.; et al. IFITM proteins drive type 2 T helper cell differentiation and exacerbate allergic airway inflammation. Eur. J. Immunol. 2019, 49, 66–78. [Google Scholar] [CrossRef]
- Tareen, S.U.; Emerman, M. Human Trim5α has additional activities that are uncoupled from retroviral capsid recognition. Virology 2011, 409, 113–120. [Google Scholar] [CrossRef]
- Yudina, Z.; Roa, A.; Johnson, R.; Biris, N.; de Souza Aranha Vieira, D.A.; Tsiperson, V.; Reszka, N.; Taylor, A.B.; Hart, P.J.; Demeler, B.; et al. RING dimerization links higher-order assembly of TRIM5α to synthesis of K63-linked polyubiquitin. Cell Rep. 2015, 12, 788–797. [Google Scholar] [CrossRef]
- Nepveu-Traversy, M.-É.; Berthoux, L. The conserved sumoylation consensus site in TRIM5α modulates its immune activation functions. Virus Res. 2014, 184, 30–38. [Google Scholar] [CrossRef]
- Dutrieux, J.; Portilho, D.M.; Arhel, N.J.; Hazan, U.; Nisole, S. TRIM5α is a SUMO substrate. Retrovirology 2015, 12, 28. [Google Scholar] [CrossRef]
- Versteeg, G.A.; Rajsbaum, R.; Sánchez-Aparicio, M.T.; Maestre, A.M.; Valdiviezo, J.; Shi, M.; Inn, K.-S.; Fernandez-Sesma, A.; Jung, J.; García-Sastre, A. The E3-ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity 2013, 38, 384–398. [Google Scholar] [CrossRef]
- Uchil, P.D.; Hinz, A.; Siegel, S.; Coenen-Stass, A.; Pertel, T.; Luban, J.; Mothes, W. TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J. Virol. 2013, 87, 257–272. [Google Scholar] [CrossRef]
- Portilho, D.M.; Fernandez, J.; Ringeard, M.; Machado, A.K.; Boulay, A.; Mayer, M.; Müller-Trutwin, M.; Beignon, A.-S.; Kirchhoff, F.; Nisole, S.; et al. Endogenous TRIM5α function is regulated by SUMOylation and nuclear sequestration for efficient innate sensing in dendritic cells. Cell Rep. 2016, 14, 355–369. [Google Scholar] [CrossRef]
- Jimenez-Moyano, E.; Ruiz, A.; Kløverpris, H.N.; Rodriguez-Plata, M.T.; Peña, R.; Blondeau, C.; Selwood, D.L.; Izquierdo-Useros, N.; Moris, A.; Clotet, B.; et al. Nonhuman TRIM5 variants enhance recognition of HIV-1-infected cells by CD8+ T cells. J. Virol. 2016, 90, 8552–8562. [Google Scholar] [CrossRef]
- Norman, J.M.; Mashiba, M.; McNamara, L.A.; Onafuwa-Nuga, A.; Chiari-Fort, E.; Shen, W.; Collins, K.L. The antiviral factor APOBEC3G enhances the recognition of HIV-infected primary T cells by natural killer cells. Nat. Immunol. 2011, 12, 975–983. [Google Scholar] [CrossRef] [Green Version]
- Casartelli, N.; Guivel-Benhassine, F.; Bouziat, R.; Brandler, S.; Schwartz, O.; Moris, A. The antiviral factor APOBEC3G improves CTL recognition of cultured HIV-infected T cells. J. Exp. Med. 2010, 207, 39–49. [Google Scholar] [CrossRef]
- Cardinaud, S.; Urrutia, A.; Rouers, A.; Coulon, P.-G.; Kervevan, J.; Richetta, C.; Bet, A.; Maze, E.A.; Larsen, M.; Iglesias, M.-C.; et al. Triggering of TLR-3, -4, NOD2, and DC-SIGN reduces viral replication and increases T-cell activation capacity of HIV-infected human dendritic cells. Eur. J. Immunol. 2017, 47, 818–829. [Google Scholar] [CrossRef] [Green Version]
- Monajemi, M.; Woodworth, C.F.; Zipperlen, K.; Gallant, M.; Grant, M.D.; Larijani, M. Positioning of APOBEC3G/F mutational hotspots in the human immunodeficiency virus genome favors reduced recognition by CD8+ T cells. PLoS ONE 2014, 9, e93428. [Google Scholar] [CrossRef]
- Grant, M.; Larijani, M. Evasion of adaptive immunity by HIV through the action of host APOBEC3G/F enzymes. AIDS Res. Ther. 2017, 14. [Google Scholar] [CrossRef]
- Tokarev, A.; Suarez, M.; Kwan, W.; Fitzpatrick, K.; Singh, R.; Guatelli, J. Stimulation of NF-κB activity by the HIV restriction factor BST2. J. Virol. 2013, 87, 2046–2057. [Google Scholar] [CrossRef]
- Galão, R.P.; Le Tortorec, A.; Pickering, S.; Kueck, T.; Neil, S.J.D. Innate sensing of HIV-1 assembly by Tetherin induces NFκB-dependent proinflammatory responses. Cell Host Microbe 2012, 12, 633–644. [Google Scholar] [CrossRef]
- Sauter, D.; Hotter, D.; Van Driessche, B.; Stürzel, C.M.; Kluge, S.F.; Wildum, S.; Yu, H.; Baumann, B.; Wirth, T.; Plantier, J.-C.; et al. Differential regulation of NF-κB-mediated proviral and antiviral host gene expression by primate lentiviral Nef and Vpu proteins. Cell Rep. 2015, 10, 586–599. [Google Scholar] [CrossRef]
- Li, S.X.; Barrett, B.S.; Heilman, K.J.; Messer, R.J.; Liberatore, R.A.; Bieniasz, P.D.; Kassiotis, G.; Hasenkrug, K.J.; Santiago, M.L. Tetherin promotes the innate and adaptive cell-mediated immune response against retrovirus infection in vivo. J. Immunol. 2014, 193, 306–316. [Google Scholar] [CrossRef]
- Li, S.X.; Barrett, B.S.; Guo, K.; Kassiotis, G.; Hasenkrug, K.J.; Dittmer, U.; Gibbert, K.; Santiago, M.L. Tetherin/BST-2 promotes dendritic cell activation and function during acute retrovirus infection. Sci. Rep. 2016, 6, 20425. [Google Scholar] [CrossRef] [Green Version]
- Arias, J.F.; Heyer, L.N.; von Bredow, B.; Weisgrau, K.L.; Moldt, B.; Burton, D.R.; Rakasz, E.G.; Evans, D.T. Tetherin antagonism by Vpu protects HIV-infected cells from antibody-dependent cell-mediated cytotoxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 6425–6430. [Google Scholar] [CrossRef] [Green Version]
- Richard, J.; Prévost, J.; von Bredow, B.; Ding, S.; Brassard, N.; Medjahed, H.; Coutu, M.; Melillo, B.; Bibollet-Ruche, F.; Hahn, B.H.; et al. BST-2 expression modulates small CD4-mimetic sensitization of HIV-1-infected cells to antibody-dependent cellular cytotoxicity. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Prévost, J.; Pickering, S.; Mumby, M.J.; Medjahed, H.; Gendron-Lepage, G.; Delgado, G.G.; Dirk, B.S.; Dikeakos, J.D.; Stürzel, C.M.; Sauter, D.; et al. Upregulation of BST-2 by type I interferons reduces the capacity of vpu to protect HIV-1-infected cells from NK cell responses. mBio 2019, 10. [Google Scholar] [CrossRef]
- Lee, H.; Komano, J.; Saitoh, Y.; Yamaoka, S.; Kozaki, T.; Misawa, T.; Takahama, M.; Satoh, T.; Takeuchi, O.; Yamamoto, N.; et al. Zinc-finger antiviral protein mediates retinoic acid inducible gene I-like receptor-independent antiviral response to murine leukemia virus. Proc. Natl. Acad. Sci. USA 2013, 110, 12379–12384. [Google Scholar] [CrossRef]
- Shenoy, A.R.; Wellington, D.A.; Kumar, P.; Kassa, H.; Booth, C.J.; Cresswell, P.; MacMicking, J.D. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science 2012, 336, 481–485. [Google Scholar] [CrossRef]
- Meunier, E.; Wallet, P.; Dreier, R.F.; Costanzo, S.; Anton, L.; Rühl, S.; Dussurgey, S.; Dick, M.S.; Kistner, A.; Rigard, M.; et al. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat. Immunol. 2015, 16, 476–484. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Cao, Z.; Wang, L.; Wan, Y.; Peng, N.; Wang, Q.; Chen, X.; Zhou, Y.; Zhu, Y. Inducible GBP5 mediates the antiviral response via interferon-related pathways during influenza a virus infection. J. Innate. Immun. 2017, 9, 419–435. [Google Scholar] [CrossRef]
- Rice, G.I.; Bond, J.; Asipu, A.; Brunette, R.L.; Manfield, I.W.; Carr, I.M.; Fuller, J.C.; Jackson, R.M.; Lamb, T.; Briggs, T.A.; et al. Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat. Genet. 2009, 41, 829–832. [Google Scholar] [CrossRef]
- Antonucci, J.M.; St Gelais, C.; Wu, L. The dynamic interplay between HIV-1, SAMHD1, and the innate antiviral response. Front. Immunol. 2017, 8, 1541. [Google Scholar] [CrossRef]
- Puigdomènech, I.; Casartelli, N.; Porrot, F.; Schwartz, O. SAMHD1 restricts HIV-1 cell-to-cell transmission and limits immune detection in monocyte-derived dendritic cells. J. Virol. 2013, 87, 2846–2856. [Google Scholar] [CrossRef]
- Chen, S.; Bonifati, S.; Qin, Z.; St Gelais, C.; Kodigepalli, K.M.; Barrett, B.S.; Kim, S.H.; Antonucci, J.M.; Ladner, K.J.; Buzovetsky, O.; et al. SAMHD1 suppresses innate immune responses to viral infections and inflammatory stimuli by inhibiting the NF-κB and interferon pathways. Proc. Natl. Acad. Sci. USA 2018, 115, E3798–E3807. [Google Scholar] [CrossRef]
- Ayinde, D.; Bruel, T.; Cardinaud, S.; Porrot, F.; Prado, J.G.; Moris, A.; Schwartz, O. SAMHD1 limits HIV-1 antigen presentation by monocyte-derived dendritic cells. J. Virol. 2015, 89, 6994–7006. [Google Scholar] [CrossRef]
- Cao, W.; Bover, L.; Cho, M.; Wen, X.; Hanabuchi, S.; Bao, M.; Rosen, D.B.; Wang, Y.-H.; Shaw, J.L.; Du, Q.; et al. Regulation of TLR7/9 responses in plasmacytoid dendritic cells by BST2 and ILT7 receptor interaction. J. Exp. Med. 2009, 206, 1603–1614. [Google Scholar] [CrossRef]
- Bego, M.G.; Côté, É.; Aschman, N.; Mercier, J.; Weissenhorn, W.; Cohen, É.A. Vpu exploits the cross-talk between BST2 and the ILT7 receptor to suppress anti-HIV-1 responses by plasmacytoid dendritic cells. PLoS Pathog. 2015, 11, e1005024. [Google Scholar] [CrossRef]
- Jin, S.; Tian, S.; Luo, M.; Xie, W.; Liu, T.; Duan, T.; Wu, Y.; Cui, J. Tetherin suppresses type I interferon signaling by targeting MAVS for NDP52-mediated selective autophagic degradation in human cells. Mol. Cell 2017, 68, 308–322.e4. [Google Scholar] [CrossRef]
- Wu, T.; Ma, F.; Ma, X.; Jia, W.; Pan, E.; Cheng, G.; Chen, L.; Sun, C. Regulating innate and adaptive immunity for controlling SIV infection by 25-hydroxycholesterol. Front. Immunol. 2018, 9, 2686. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Restriction Factor | Induction by Innate Immunity | Antiviral Function | Feedback on Immunity | HIV Counteraction |
|---|---|---|---|---|
| IFITM1/2/3 | IFN-α IFN-γ | - Inhibit viral entry by reducing membrane fluidity - Negative imprinting of virions | - Involved in Th1/Th2 polarization of CD4+ T cells - Targeting of virus in endosomes for TLRs sensing? | |
| TRIM5α | IFN-I | - Premature uncoating - Targets viral capsid for proteasomal degradation | - Activation of NF-κB after sensing the viral capsid | Viral capsids with reduced affinity for TRIM5α |
| APOBEC3G | IFN-α IFN-γ IL-2, IL17, IL15 | - Deamination of cytidines to uracils during RT creating hypermutated proviral DNA | - Induction of NKG2D ligand expression leading to the recognition of infected cells by NK cells - Generation of antigenic peptides presented on MHC-I allowing the recognition of infected cells by CD8 T cells | HIV-1 Vif |
| SAMHD1 | TLR3, RIG-I and MDA5 activation (in HeLa, HEK293 and MARC-145 cells) IFN-α (liver cells) IL-12, IL18 | - Inhibits RT by decreasing the cellular pool of dNTPs - Degradation of HIV genomic RNA | - Inhibition of NF-κB - Decreases antigen presentation | HIV-2 Vpx |
| Mx2 | IFN-α, IFN-β IFN-γ | - Inhibits HIV nuclear import - Impairs uncoating? | Not described | |
| BST-2/Tetherin | IFN-α IFN-γ IL27 | - HIV budding (viral entrapment) - Internalization and degradation of virions by the endosomal pathway | - Activation of NF-κB - Delivery of PAMPs on endosomal TLRs? - Sensitizes infected cells to ADCC - Sensitizes infected cells to NK cells - Binds to ILT7 on pDCs and inhibits IFN-I production - Inhibits RIG-I signaling (leads to the autophagic degradation of MAVS) | HIV-1 Vpu |
| CH25 | IFN-α, IFN-β IFN-γ TLR4/TLR3 activation | - Inhibits viral entry by affecting membrane fluidity | - Inhibits inflammation - Blocks Ig class switch recombination in B cells | |
| ZAP | IFN-α | - Degradation of viral mRNA - Inhibition of translation | - ZAP-S promotes RIG-I activity | HIV-Rev via Matrin3 |
| SLFN11 | IFN-I | - Inhibits HIV translation by preventing the change of tRNA pool composition | Not described | |
| ISG15 | IFN-α, IFN-β NF-κB | - Inhibits HIV-1 release (ISGylation of Gag) | Not described | |
| GBP5 | IFN-I | - Decreases viral progeny infectivity by impairing incorporation of gp120 in budding viruses | - Activates NLRP3 and AIM2 inflammasomes - Stimulation of NF-κB |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergantz, L.; Subra, F.; Deprez, E.; Delelis, O.; Richetta, C. Interplay between Intrinsic and Innate Immunity during HIV Infection. Cells 2019, 8, 922. https://doi.org/10.3390/cells8080922
Bergantz L, Subra F, Deprez E, Delelis O, Richetta C. Interplay between Intrinsic and Innate Immunity during HIV Infection. Cells. 2019; 8(8):922. https://doi.org/10.3390/cells8080922
Chicago/Turabian StyleBergantz, Louis, Frédéric Subra, Eric Deprez, Olivier Delelis, and Clémence Richetta. 2019. "Interplay between Intrinsic and Innate Immunity during HIV Infection" Cells 8, no. 8: 922. https://doi.org/10.3390/cells8080922
APA StyleBergantz, L., Subra, F., Deprez, E., Delelis, O., & Richetta, C. (2019). Interplay between Intrinsic and Innate Immunity during HIV Infection. Cells, 8(8), 922. https://doi.org/10.3390/cells8080922