A Novel Chromosomal Translocation Identified due to Complex Genetic Instability in iPSC Generated for Choroideremia

 , ,
, , 
Abstract
:1. Background
2. Materials and Methods
2.1. Genetic and Clinical Investigations
2.2. Skin Biopsy and Fibroblast Culture
2.3. Reprogramming of iPSCs
2.4. In Vitro Differentiation Assay
2.5. Differentiation of iPSC-Derived RPE
2.6. qPCR Analysis
2.7. Western Blot Analysis
2.8. Karyotype Analysis
2.9. Copy Number Variant Analysis
2.10. Immunofluorescence Studies
2.11. Transmission Electron Microscopy
2.12. Transepithelial Resistance Measurements
2.13. Phagocytosis Assay
2.14. In Vitro Prenylation Assay
2.15. Statistical Analysis
3. Results
3.1. Typical Choroideremia Clinical Phenotype
3.2. Lack of REP1 Production in the iPSCs of CHM4 and CHM5
3.3. High Level of Genetic Instability in the iPSCs of CHM4 and CHM5
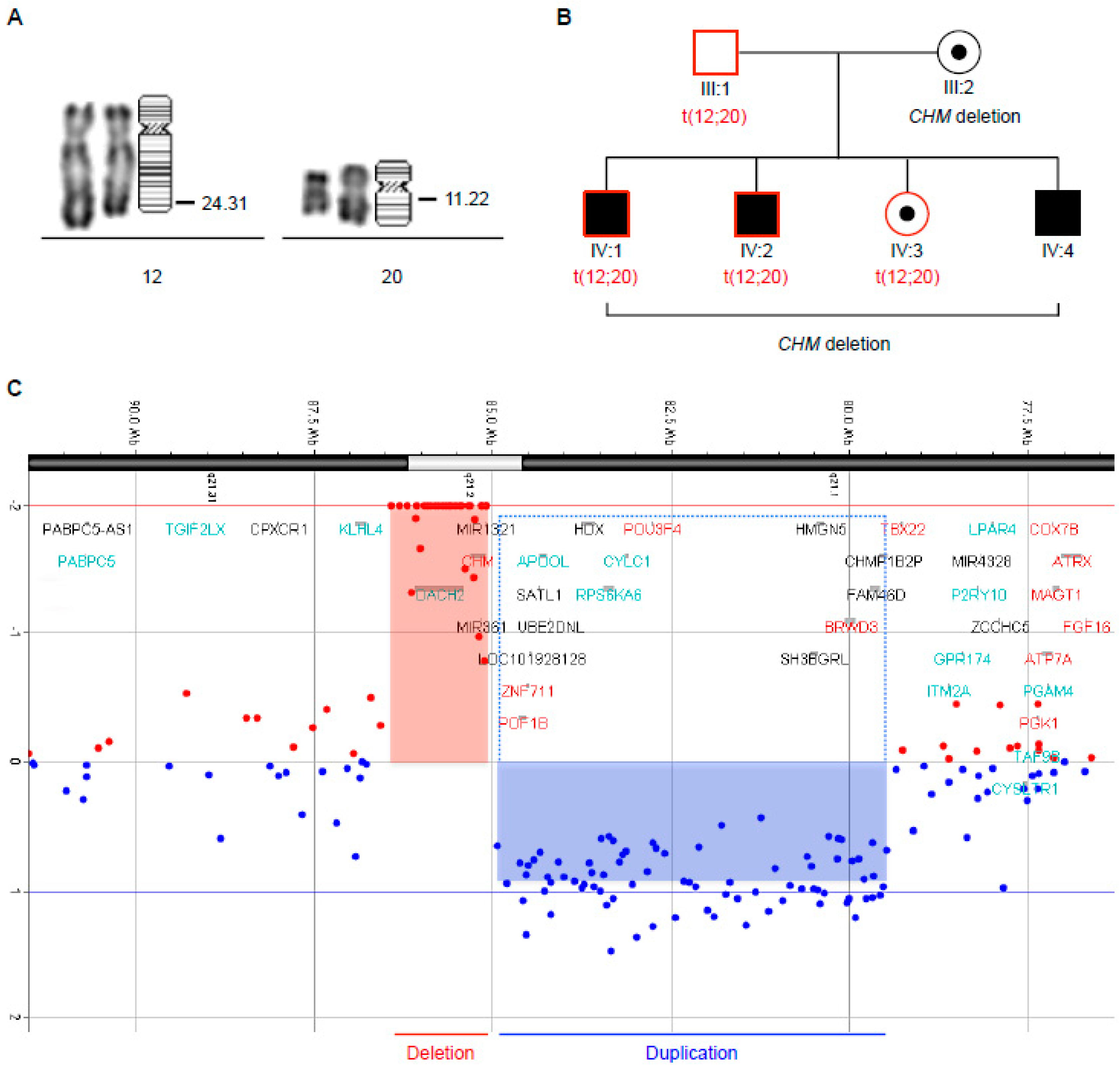
3.4. Familial Segregation of a Novel Chromosomal Translocation
3.5. CHM Retinal Phenotype Is Not Further Impacted by the Translocation or Duplication
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grimm, S. The art and design of genetic screens: Mammalian culture cells. Nat. Rev. Genet. 2004, 5, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Thomson, J.A. Pluripotent stem cell lines. Genes Dev. 2008, 22, 1987–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Trokovic, R.; Weltner, J.; Nishimura, K.; Ohtaka, M.; Nakanishi, M.; Salomaa, V.; Jalanko, A.; Otonkoski, T.; Kyttala, A. Advanced feeder-free generation of induced pluripotent stem cells directly from blood cells. Stem Cells Transl. Med. 2014, 3, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, S.; Goldstein, R.A.; Nierras, C.R. The promise of human induced pluripotent stem cells for research and therapy. Nat. Rev. Mol. Cell Biol. 2008, 9, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Sanjurjo-Soriano, C.; Kalatzis, V. Guiding Lights in Genome Editing for Inherited Retinal Disorders: Implications for Gene and Cell Therapy. Neural Plast. 2018, 5056279. [Google Scholar] [CrossRef]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef]
- Nakano, T.; Ando, S.; Takata, N.; Kawada, M.; Muguruma, K.; Sekiguchi, K.; Saito, K.; Yonemura, S.; Eiraku, M.; Sasai, Y. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 2012, 10, 771–785. [Google Scholar] [CrossRef]
- Hirami, Y.; Osakada, F.; Takahashi, K.; Okita, K.; Yamanaka, S.; Ikeda, H.; Yoshimura, N.; Takahashi, M. Generation of retinal cells from mouse and human induced pluripotent stem cells. Neurosci. Lett. 2009, 458, 126–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, J.S.; Shearer, R.L.; Capowski, E.E.; Wright, L.S.; Wallace, K.A.; McMillan, E.L.; Zhang, S.C.; Gamm, D.M. Modeling early retinal development with human embryonic and induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 16698–16703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cereso, N.; Pequignot, M.O.; Robert, L.; Becker, F.; De Luca, V.; Nabholz, N.; Rigau, V.; De Vos, J.; Hamel, C.P.; Kalatzis, V. Proof of concept for AAV2/5-mediated gene therapy in iPSc-derived retinal pigment epithelium of choroideremia patients. Mol. Ther. Methods Clin. Dev. 2014, 1, 14011. [Google Scholar] [CrossRef] [PubMed]
- Burnight, E.R.; Wiley, L.A.; Drack, A.V.; Braun, T.A.; Anfinson, K.R.; Kaalberg, E.E.; Halder, J.A.; Affatigato, L.M.; Mullins, R.F.; Stone, E.M.; et al. CEP290 gene transfer rescues Leber congenital amaurosis cellular phenotype. Gene Ther. 2014, 21, 662–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wu, W.H.; Hsu, C.W.; Nguyen, H.V.; Tsai, Y.T.; Chan, L.; Nagasaki, T.; Maumenee, I.H.; Yannuzzi, L.A.; Hoang, Q.V.; et al. Gene therapy in patient-specific stem cell lines and a preclinical model of retinitis pigmentosa with membrane frizzled-related protein defects. Mol. Ther. 2014, 22, 1688–1697. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kuai, D.; Guziewicz, K.E.; Meyer, J.; Wilson, M.; Lu, J.; Smith, M.; Clark, E.; Verhoeven, A.; Aguirre, G.D.; et al. Pharmacological Modulation of Photoreceptor Outer Segment Degradation in a Human iPS Cell Model of Inherited Macular Degeneration. Mol. Ther. 2015, 23, 1700–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, N.; Carr, A.J.; Lane, A.; Moeller, F.; Chen, L.L.; Aguila, M.; Nommiste, B.; Muthiah, M.N.; Kanuga, N.; Wolfrum, U.; et al. Translational read-through of the RP2 Arg120stop mutation in patient iPSC-derived retinal pigment epithelium cells. Hum. Mol. Genet. 2015, 24, 972–986. [Google Scholar] [CrossRef]
- Ramsden, C.M.; Nommiste, B.; Lane, A.R.; Carr, A.F.; Powner, M.B.; Smart, M.J.K.; Chen, L.L.; Muthiah, M.N.; Webster, A.R.; Moore, A.T.; et al. Rescue of the MERTK phagocytic defect in a human iPSC disease model using translational read-through inducing drugs. Sci. Rep. 2017, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Torriano, S.; Erkilic, N.; Faugere, V.; Damodar, K.; Hamel, C.P.; Roux, A.F.; Kalatzis, V. Pathogenicity of a novel missense variant associated with choroideremia and its impact on gene replacement therapy. Hum. Mol. Genet. 2017, 26, 3573–3584. [Google Scholar] [CrossRef]
- Torriano, S.; Erkilic, N.; Baux, D.; Cereso, N.; De Luca, V.; Meunier, I.; Moosajee, M.; Roux, A.F.; Hamel, C.P.; Kalatzis, V. The effect of PTC124 on choroideremia fibroblasts and iPSC-derived RPE raises considerations for therapy. Sci. Rep. 2018, 8, 8234. [Google Scholar] [CrossRef]
- Seabra, M.C.; Brown, M.S.; Slaughter, C.A.; Sudhof, T.C.; Goldstein, J.L. Purification of component A of Rab geranylgeranyl transferase: Possible identity with the choroideremia gene product. Cell 1992, 70, 1049–1057. [Google Scholar] [CrossRef]
- Andres, D.A.; Seabra, M.C.; Brown, M.S.; Armstrong, S.A.; Smeland, T.E.; Cremers, F.P.; Goldstein, J.L. cDNA cloning of component A of Rab geranylgeranyl transferase and demonstration of its role as a Rab escort protein. Cell 1993, 73, 1091–1099. [Google Scholar] [CrossRef]
- Starr, C.J.; Kappler, J.A.; Chan, D.K.; Kollmar, R.; Hudspeth, A.J. Mutation of the zebrafish choroideremia gene encoding Rab escort protein 1 devastates hair cells. Proc. Natl. Acad. Sci. USA 2004, 101, 2572–2577. [Google Scholar] [CrossRef] [PubMed]
- van den Hurk, J.A.; Hendriks, W.; van de Pol, D.J.; Oerlemans, F.; Jaissle, G.; Ruther, K.; Kohler, K.; Hartmann, J.; Zrenner, E.; van Bokhoven, H.; et al. Mouse choroideremia gene mutation causes photoreceptor cell degeneration and is not transmitted through the female germline. Hum. Mol. Genet. 1997, 6, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Tolmachova, T.; Anders, R.; Abrink, M.; Bugeon, L.; Dallman, M.J.; Futter, C.E.; Ramalho, J.S.; Tonagel, F.; Tanimoto, N.; Seeliger, M.W.; et al. Independent degeneration of photoreceptors and retinal pigment epithelium in conditional knockout mouse models of choroideremia. J. Clin. Investig. 2006, 116, 386–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duong, T.T.; Vasireddy, V.; Ramachandran, P.; Herrera, P.S.; Leo, L.; Merkel, C.; Bennett, J.; Mills, J.A. Use of induced pluripotent stem cell models to probe the pathogenesis of Choroideremia and to develop a potential treatment. Stem Cell Res. 2018, 27, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Weissbein, U.; Benvenisty, N.; Ben-David, U. Quality control: Genome maintenance in pluripotent stem cells. J. Cell Biol. 2014, 204, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Gomez-Gonzalez, B. Genome instability: A mechanistic view of its causes and consequences. Nat. Rev. Genet. 2008, 9, 204–217. [Google Scholar] [CrossRef]
- Sanchez-Alcudia, R.; Garcia-Hoyos, M.; Lopez-Martinez, M.A.; Sanchez-Bolivar, N.; Zurita, O.; Gimenez, A.; Villaverde, C.; Rodrigues-Jacy da Silva, L.; Corton, M.; Perez-Carro, R.; et al. A Comprehensive Analysis of Choroideremia: From Genetic Characterization to Clinical Practice. PLoS ONE 2016, 11, e0151943. [Google Scholar] [CrossRef]
- Ramirez, J.M.; Bai, Q.; Pequignot, M.; Becker, F.; Kassambara, A.; Bouin, A.; Kalatzis, V.; Dijon-Grinand, M.; De Vos, J. Side scatter intensity is highly heterogeneous in undifferentiated pluripotent stem cells and predicts clonogenic self-renewal. Stem Cells Dev. 2013, 22, 1851–1860. [Google Scholar] [CrossRef]
- Moralli, D.; Yusuf, M.; Mandegar, M.A.; Khoja, S.; Monaco, Z.L.; Volpi, E.V. An improved technique for chromosomal analysis of human ES and iPS cells. Stem Cell Rev. 2011, 7, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Hoyos, M.; Lorda-Sanchez, I.; Gomez-Garre, P.; Villaverde, C.; Cantalapiedra, D.; Bustamante, A.; Diego-Alvarez, D.; Vallespin, E.; Gallego-Merlo, J.; Trujillo, M.J.; et al. New type of mutations in three spanish families with choroideremia. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Vache, C.; Torriano, S.; Faugere, V.; Erkilic, N.; Baux, D.; Garcia-Garcia, G.; Hamel, C.P.; Meunier, I.; Zanlonghi, X.; Koenig, M.; et al. Pathogenicity of novel atypical variants leading to choroideremia as determined by functional analyses. Hum. Mutat. 2019, 40, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Radziwon, A.; Arno, G.; Wheaton, K.D.; McDonagh, E.M.; Baple, E.L.; Webb-Jones, K.; Birch, G.D.; Webster, A.R.; MacDonald, I.M. Single-base substitutions in the CHM promoter as a cause of choroideremia. Hum. Mutat. 2017, 38, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J.; Pesah, Y.I.; Harding, M.; Paylor, R.; Mardon, G. Mouse Dach2 mutants do not exhibit gross defects in eye development or brain function. Genesis 2006, 44, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Bione, S.; Rizzolio, F.; Sala, C.; Ricotti, R.; Goegan, M.; Manzini, M.C.; Battaglia, R.; Marozzi, A.; Vegetti, W.; Dalpra, L.; et al. Mutation analysis of two candidate genes for premature ovarian failure, DACH2 and POF1B. Hum. Reprod. 2004, 19, 2759–2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, Y.; Li, Y.; Duan, Y.; Zhang, K.; Wang, J.; Dai, Y. Exome sequencing identifies mutations in ABCD1 and DACH2 in two brothers with a distinct phenotype. BMC Med. Genet. 2014, 15, 105. [Google Scholar] [CrossRef] [PubMed]
- van der Werf, I.M.; Van Dijck, A.; Reyniers, E.; Helsmoortel, C.; Kumar, A.A.; Kalscheuer, V.M.; de Brouwer, A.P.; Kleefstra, T.; van Bokhoven, H.; Mortier, G.; et al. Mutations in two large pedigrees highlight the role of ZNF711 in X-linked intellectual disability. Gene 2017, 605, 92–98. [Google Scholar] [CrossRef]
- Field, M.; Tarpey, P.S.; Smith, R.; Edkins, S.; O’Meara, S.; Stevens, C.; Tofts, C.; Teague, J.; Butler, A.; Dicks, E.; et al. Mutations in the BRWD3 gene cause X-linked mental retardation associated with macrocephaly. Am. J. Hum. Genet. 2007, 81, 367–374. [Google Scholar] [CrossRef]
- Grotto, S.; Drouin-Garraud, V.; Ounap, K.; Puusepp-Benazzouz, H.; Schuurs-Hoeijmakers, J.; Le Meur, N.; Chambon, P.; Fehrenbach, S.; van Bokhoven, H.; Frebourg, T.; et al. Clinical assessment of five patients with BRWD3 mutation at Xq21.1 gives further evidence for mild to moderate intellectual disability and macrocephaly. Eur. J. Med. Genet. 2014, 57, 200–206. [Google Scholar] [CrossRef]
- Yntema, H.G.; van den Helm, B.; Kissing, J.; van Duijnhoven, G.; Poppelaars, F.; Chelly, J.; Moraine, C.; Fryns, J.P.; Hamel, B.C.; Heilbronner, H.; et al. A novel ribosomal S6-kinase (RSK4; RPS6KA6) is commonly deleted in patients with complex X-linked mental retardation. Genomics 1999, 62, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Okten, G.; Gunes, S.; Onat, O.E.; Tukun, A.; Ozcelik, T.; Kocak, I. Disruption of HDX gene in premature ovarian failure. Syst. Biol. Reprod. Med. 2013, 59, 218–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacombe, A.; Lee, H.; Zahed, L.; Choucair, M.; Muller, J.M.; Nelson, S.F.; Salameh, W.; Vilain, E. Disruption of POF1B binding to nonmuscle actin filaments is associated with premature ovarian failure. Am. J. Hum. Genet. 2006, 79, 113–119. [Google Scholar] [CrossRef] [PubMed]
- de Kok, Y.J.; van der Maarel, S.M.; Bitner-Glindzicz, M.; Huber, I.; Monaco, A.P.; Malcolm, S.; Pembrey, M.E.; Ropers, H.H.; Cremers, F.P. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science 1995, 267, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Bitner-Glindzicz, M.; Turnpenny, P.; Hoglund, P.; Kaariainen, H.; Sankila, E.M.; van der Maarel, S.M.; de Kok, Y.J.; Ropers, H.H.; Cremers, F.P.; Pembrey, M.; et al. Further mutations in Brain 4 (POU3F4) clarify the phenotype in the X-linked deafness, DFN3. Hum. Mol. Genet. 1995, 4, 1467–1469. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Hoyos, M.; Sanz, R.; Diego-Alvarez, D.; Lorda-Sanchez, I.; Trujillo-Tiebas, M.J.; Cantalapiedra, D.; Ramos, C.; Ayuso, C. New approach for the refinement of the location of the X-chromosome breakpoint in a previously described female patient with choroideremia carrying a X;4 translocation. Am. J. Med. Genet. Part A 2005, 138, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Lorda-Sanchez, I.J.; Ibanez, A.J.; Sanz, R.J.; Trujillo, M.J.; Anabitarte, M.E.; Querejeta, M.E.; Rodriguez de Alba, M.; Gimenez, A.; Infantes, F.; Ramos, C.; et al. Choroideremia, sensorineural deafness, and primary ovarian failure in a woman with a balanced X-4 translocation. Ophthalmic Genet. 2000, 21, 185–189. [Google Scholar] [CrossRef]
- Weber, T.A.; Koob, S.; Heide, H.; Wittig, I.; Head, B.; van der Bliek, A.; Brandt, U.; Mittelbronn, M.; Reichert, A.S. APOOL is a cardiolipin-binding constituent of the Mitofilin/MINOS protein complex determining cristae morphology in mammalian mitochondria. PLoS ONE 2013, 8, e63683. [Google Scholar] [CrossRef] [PubMed]
- Maurin, M.L.; Arfeuille, C.; Sonigo, P.; Rondeau, S.; Vekemans, M.; Turleau, C.; Ville, Y.; Malan, V. Large Duplications Can Be Benign Copy Number Variants: A Case of a 3.6-Mb Xq21.33 Duplication. Cytogenet. Genome Res. 2017, 151, 115–118. [Google Scholar] [CrossRef]
- Mackic-Djurovic, M.; Hasic, S.; Kiseljakovic, E.; Rukavina, D.; Ibrulj, S. Cytogenetic Abnormalities Found in Patients with Reproductive Problems. Med. Arch. 2017, 71, 396–399. [Google Scholar] [CrossRef] [Green Version]
- Mackie Ogilvie, C.; Scriven, P.N. Meiotic outcomes in reciprocal translocation carriers ascertained in 3-day human embryos. Eur. J. Hum. Genet. 2002, 10, 801–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzumori, N.; Sugiura-Ogasawara, M. Genetic factors as a cause of miscarriage. Curr. Med. Chem. 2010, 17, 3431–3437. [Google Scholar] [CrossRef] [PubMed]
- Redin, C.; Brand, H.; Collins, R.L.; Kammin, T.; Mitchell, E.; Hodge, J.C.; Hanscom, C.; Pillalamarri, V.; Seabra, C.M.; Abbott, M.A.; et al. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat. Genet. 2017, 49, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, B.; Samuel, C.R.; Thyagarajan, S.S. Balanced Autosomal Translocations in Two Women Reporting Recurrent Miscarriage. J. Clin. Diagn. Res. 2016, 10, GD01–GD03. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, E.; Voet, T.; Le Caignec, C.; Ampe, M.; Konings, P.; Melotte, C.; Debrock, S.; Amyere, M.; Vikkula, M.; Schuit, F.; et al. Chromosome instability is common in human cleavage-stage embryos. Nat. Med. 2009, 15, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Alfarawati, S.; Fragouli, E.; Colls, P.; Wells, D. Embryos of robertsonian translocation carriers exhibit a mitotic interchromosomal effect that enhances genetic instability during early development. PLoS Genet. 2012, 8, e1003025. [Google Scholar] [CrossRef] [PubMed]
- Cohen, O.; Mermet, M.A.; Demongeot, J. HC Forum: A web site based on an international human cytogenetic database. Nucleic Acids Res. 2001, 29, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.A.; Ghule, P.N.; Therrien, J.A.; Lian, J.B.; Stein, J.L.; van Wijnen, A.J.; Stein, G.S. Self-renewal of human embryonic stem cells is supported by a shortened G1 cell cycle phase. J. Cell Physiol. 2006, 209, 883–893. [Google Scholar] [CrossRef] [PubMed]
- International Stem Cell Initiative; Amps, K.; Andrews, P.W.; Anyfantis, G.; Armstrong, L.; Avery, S.; Baharvand, H.; Baker, J.; Baker, D.; Munoz, M.B.; et al. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat. Biotechnol. 2011, 29, 1132–1144. [Google Scholar] [Green Version]
- Lefort, N.; Feyeux, M.; Bas, C.; Feraud, O.; Bennaceur-Griscelli, A.; Tachdjian, G.; Peschanski, M.; Perrier, A.L. Human embryonic stem cells reveal recurrent genomic instability at 20q11.21. Nat. Biotechnol. 2008, 26, 1364–1366. [Google Scholar] [CrossRef] [PubMed]
- Spits, C.; Mateizel, I.; Geens, M.; Mertzanidou, A.; Staessen, C.; Vandeskelde, Y.; Van der Elst, J.; Liebaers, I.; Sermon, K. Recurrent chromosomal abnormalities in human embryonic stem cells. Nat. Biotechnol. 2008, 26, 1361–1363. [Google Scholar] [CrossRef] [PubMed]
- Mayshar, Y.; Ben-David, U.; Lavon, N.; Biancotti, J.C.; Yakir, B.; Clark, A.T.; Plath, K.; Lowry, W.E.; Benvenisty, N. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell 2010, 7, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Taapken, S.M.; Nisler, B.S.; Newton, M.A.; Sampsell-Barron, T.L.; Leonhard, K.A.; McIntire, E.M.; Montgomery, K.D. Karotypic abnormalities in human induced pluripotent stem cells and embryonic stem cells. Nat. Biotechnol. 2011, 29, 313–314. [Google Scholar] [CrossRef] [PubMed]
- Rohani, L.; Johnson, A.A.; Naghsh, P.; Rancourt, D.E.; Ulrich, H.; Holland, H. Concise Review: Molecular Cytogenetics and Quality Control: Clinical Guardians for Pluripotent Stem Cells. Stem Cells Transl. Med. 2018, 7, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Stover, A.E.; Herculian, S.; Banuelos, M.G.; Navarro, S.L.; Jenkins, M.P.; Schwartz, P.H. Culturing Human Pluripotent and Neural Stem Cells in an Enclosed Cell Culture System for Basic and Preclinical Research. J. Vis. Exp. 2016, 112, 53685. [Google Scholar] [CrossRef] [PubMed]
- Sanjurjo-Soriano, C.; Erkilic, N.; Manes, G.; Dubois, G.; Hamel, C.P.; Meunier, I.; Kalatzis, V. Generation of an iPSC line, INMi001-A, carrying the two most common USH2A mutations from a compound heterozygote with non-syndromic retinitis pigmentosa. Stem Cell Res. 2018, 33, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Sanjurjo-Soriano, C.; Erkilic, N.; Manes, G.; Dubois, G.; Hamel, C.P.; Meunier, I.; Kalatzis, V. Generation of a human iPSC line, INMi002-A, carrying the most prevalent USH2A variant associated with Usher syndrome type 2. Stem Cell Res. 2018, 33, 247–250. [Google Scholar] [CrossRef]
- Erkilic, N.; Sanjurjo-Soriano, C.; Diakatou, M.; Manes, G.; Dubois, G.; Hamel, C.P.; Meunier, I.; Kalatzis, V. Generation of a human iPSC line, INMi003-A, with a missense mutation in CRX associated with autosomal dominant cone-rod dystrophy. Stem Cell Res. 2019, 38, 101478. [Google Scholar] [CrossRef]
- Erkilic, N.; Sanjurjo-Soriano, C.; Manes, G.; Dubois, G.; Hamel, C.P.; Meunier, I.; Kalatzis, V. Generation of a human iPSC line, INMi004-A, with a point mutation in CRX associated with autosomal dominant Leber congenital amaurosis. Stem Cell Res. 2019, 38, 101476. [Google Scholar] [CrossRef]
- Salinas, S.; Erkilic, N.; Damodar, K.; Moles, J.P.; Fournier-Wirth, C.; Van de Perre, P.; Kalatzis, V.; Simonin, Y. Zika Virus Efficiently Replicates in Human Retinal Epithelium and Disturbs Its Permeability. J. Virol. 2017, 91, e02144-16. [Google Scholar] [CrossRef]
- Simonin, Y.; Erkilic, N.; Damodar, K.; Cle, M.; Desmetz, C.; Bollore, K.; Taleb, M.; Torriano, S.; Barthelemy, J.; Dubois, G.; et al. Zika virus induces strong inflammatory responses and impairs homeostasis and function of the human retinal pigment epithelium. EBioMedicine 2019, 39, 315–331. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CHM iPSC Clones | Passage | Aberrations | Consequences | |
|---|---|---|---|---|
| CHM4 | Clone 1 | P21 | 48,XY,+5,t(12;20)(q24.3;q11.2),+der(12)t(12;20)(q24.3;q11.2) | Trisomy 5, Partial trisomy 12, Partial trisomy 20 |
| Clone 2 | P19 | 47,XY,t(12;20)(q24.3;q11.2), +der(12)t(12;20) | Partial trisomy 12, Partial trisomy 20 | |
| CHM5 | Clone 1 | P13 | 47,XY, der(5)(5pter ->q12::q13.1->qter),-12,+der(12)(12qter->q24.3::q11.2->q13.1::5q12->5qter)x2, der(20)(20qter->q11.2::12q24.3->12qter) | Partial trisomy 5, Partial trisomy 12, Partial trisomy 20 |
| Clone 2 | P15 | 46,XY,t(12;20)(q24.3;q11.2) | Balanced | |
| CHM1 | Clone 1 | P17 | 46,XY,del(7)(q21) | Monosomy distal to 7q21 |
| Clone 2 | P19 | 46,XY,i(7)(p10) | Monosomy 7q, Trisomy 7p | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erkilic, N.; Gatinois, V.; Torriano, S.; Bouret, P.; Sanjurjo-Soriano, C.; De Luca, V.; Damodar, K.; Cereso, N.; Puechberty, J.; Sanchez-Alcudia, R.; et al. A Novel Chromosomal Translocation Identified due to Complex Genetic Instability in iPSC Generated for Choroideremia. Cells 2019, 8, 1068. https://doi.org/10.3390/cells8091068
Erkilic N, Gatinois V, Torriano S, Bouret P, Sanjurjo-Soriano C, De Luca V, Damodar K, Cereso N, Puechberty J, Sanchez-Alcudia R, et al. A Novel Chromosomal Translocation Identified due to Complex Genetic Instability in iPSC Generated for Choroideremia. Cells. 2019; 8(9):1068. https://doi.org/10.3390/cells8091068
Chicago/Turabian StyleErkilic, Nejla, Vincent Gatinois, Simona Torriano, Pauline Bouret, Carla Sanjurjo-Soriano, Valerie De Luca, Krishna Damodar, Nicolas Cereso, Jacques Puechberty, Rocio Sanchez-Alcudia, and et al. 2019. "A Novel Chromosomal Translocation Identified due to Complex Genetic Instability in iPSC Generated for Choroideremia" Cells 8, no. 9: 1068. https://doi.org/10.3390/cells8091068
APA StyleErkilic, N., Gatinois, V., Torriano, S., Bouret, P., Sanjurjo-Soriano, C., De Luca, V., Damodar, K., Cereso, N., Puechberty, J., Sanchez-Alcudia, R., Hamel, C. P., Ayuso, C., Meunier, I., Pellestor, F., & Kalatzis, V. (2019). A Novel Chromosomal Translocation Identified due to Complex Genetic Instability in iPSC Generated for Choroideremia. Cells, 8(9), 1068. https://doi.org/10.3390/cells8091068