splitGFP Technology Reveals Dose-Dependent ER-Mitochondria Interface Modulation by α-Synuclein A53T and A30P Mutants

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Constructs
2.2. Cell lines and Transfection
2.3. Western Blotting
2.4. Immunocytochemistry Analysis
2.5. Aequorin Measurements
2.6. ER-Mitochondria Contact Site Analysis
2.7. Statistical Analysis
3. Results
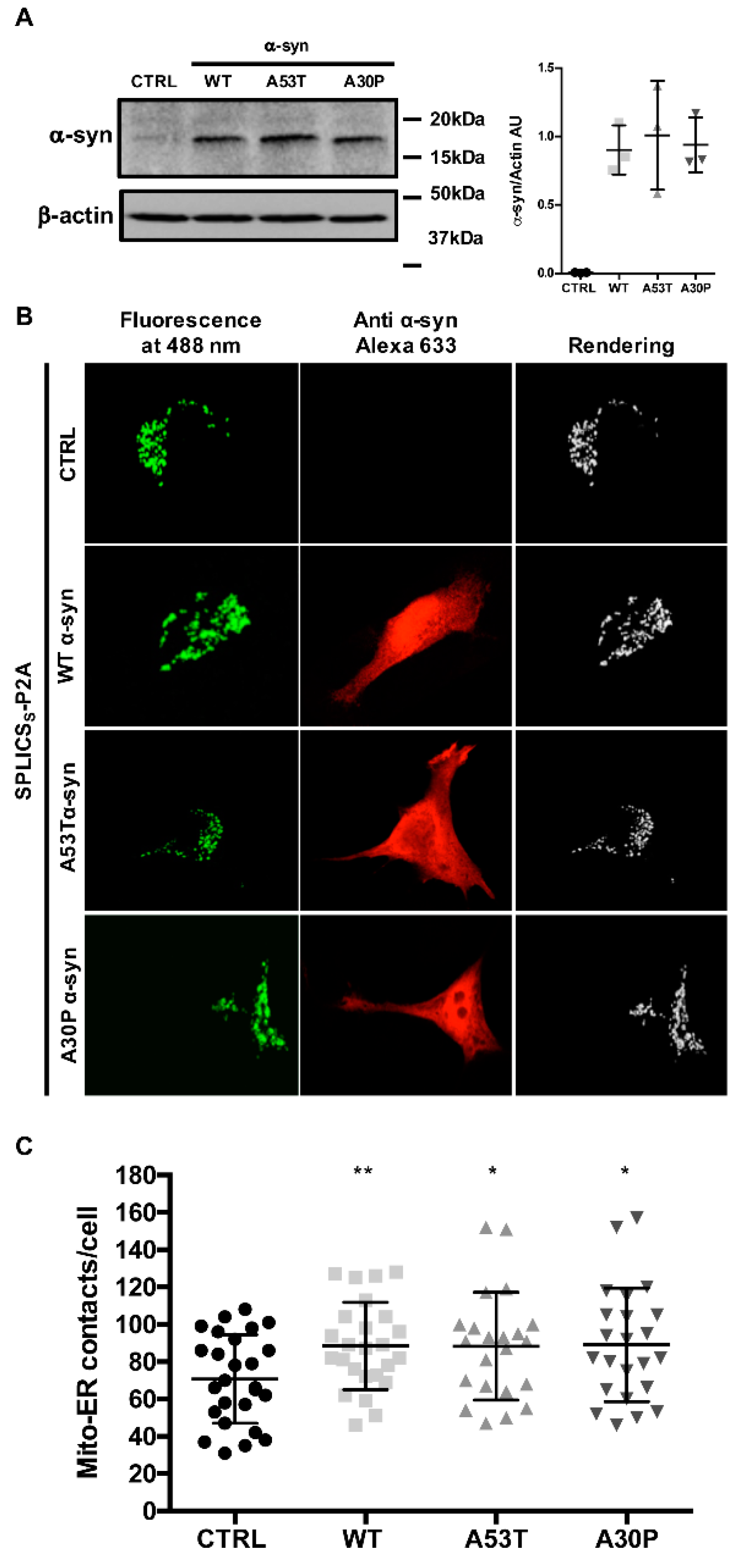
3.1. α-syn A53T and A30P Mutants Physically Modulate ER-Mitochondria Contact Sites
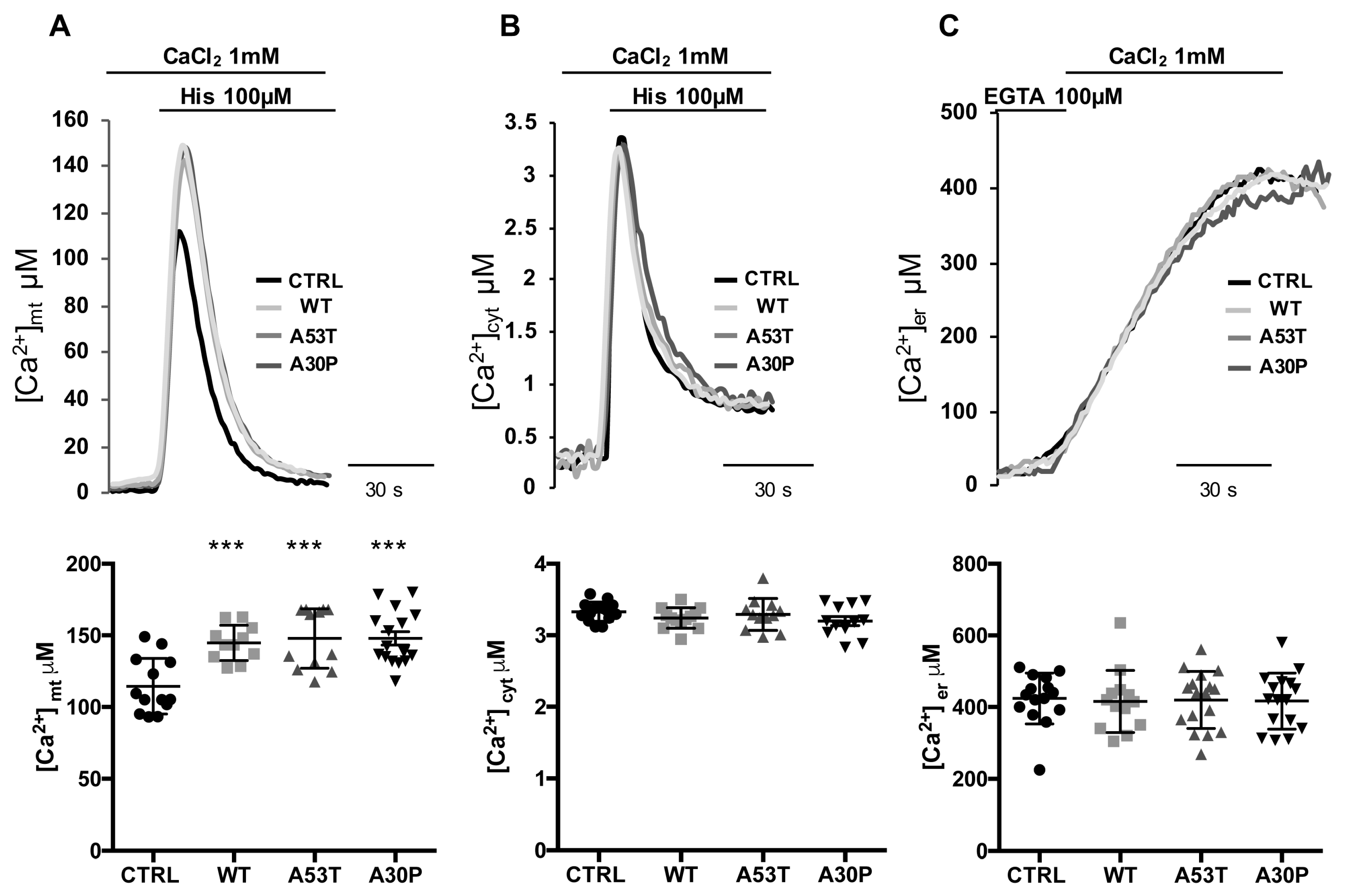
3.2. Overexpression of A53T and A30P α-Synuclein Mutants Enhances Mitochondrial Ca2+ Transients with the Same Extent than wt α-Synuclein
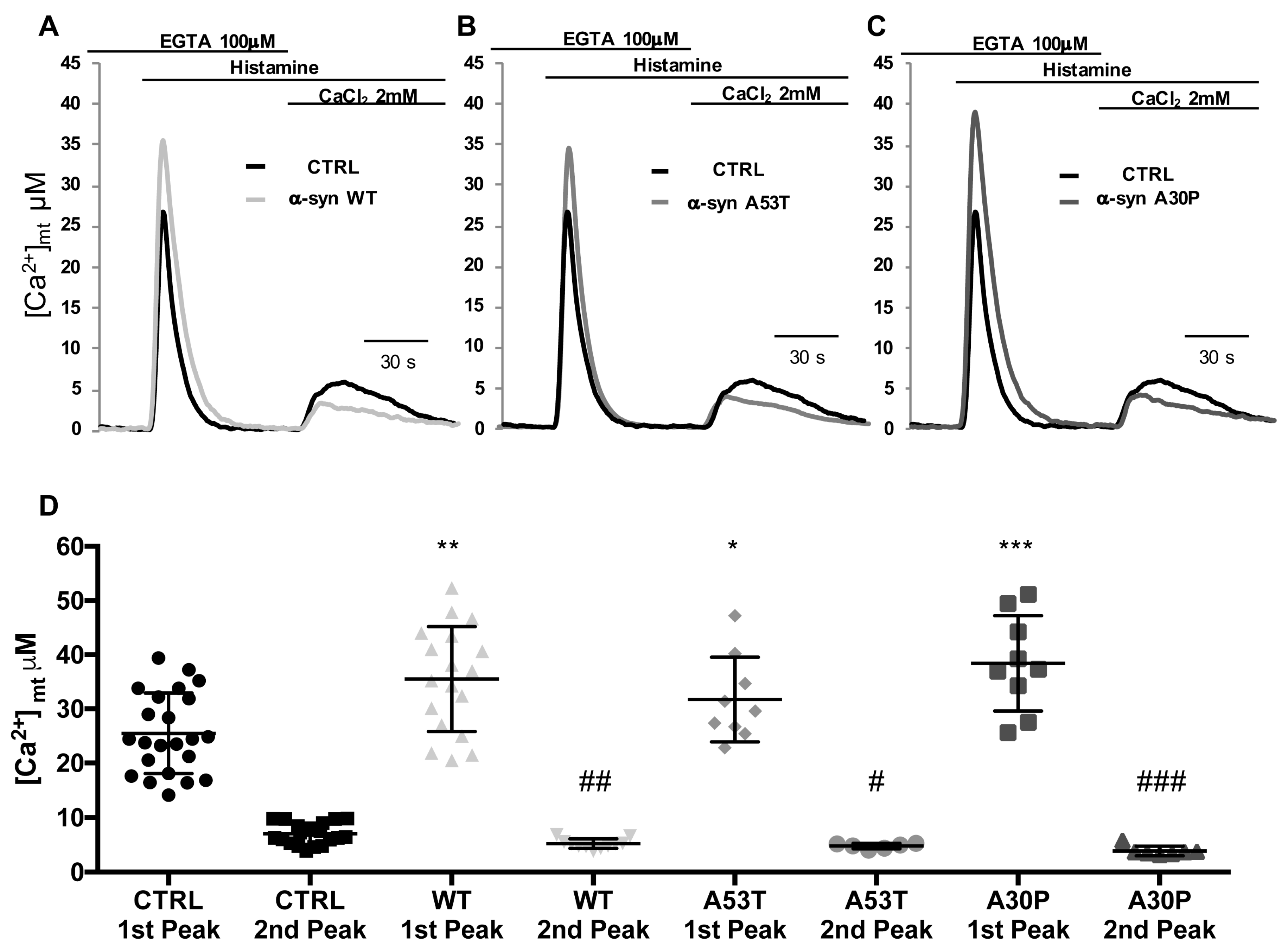
3.3. α-syn A53T and A30P Mutants Enhance ER-Mitochondria Ca2+ Transfer but Impair Mitochondrial Ca2+ Uptake from the Extracellular Milieu
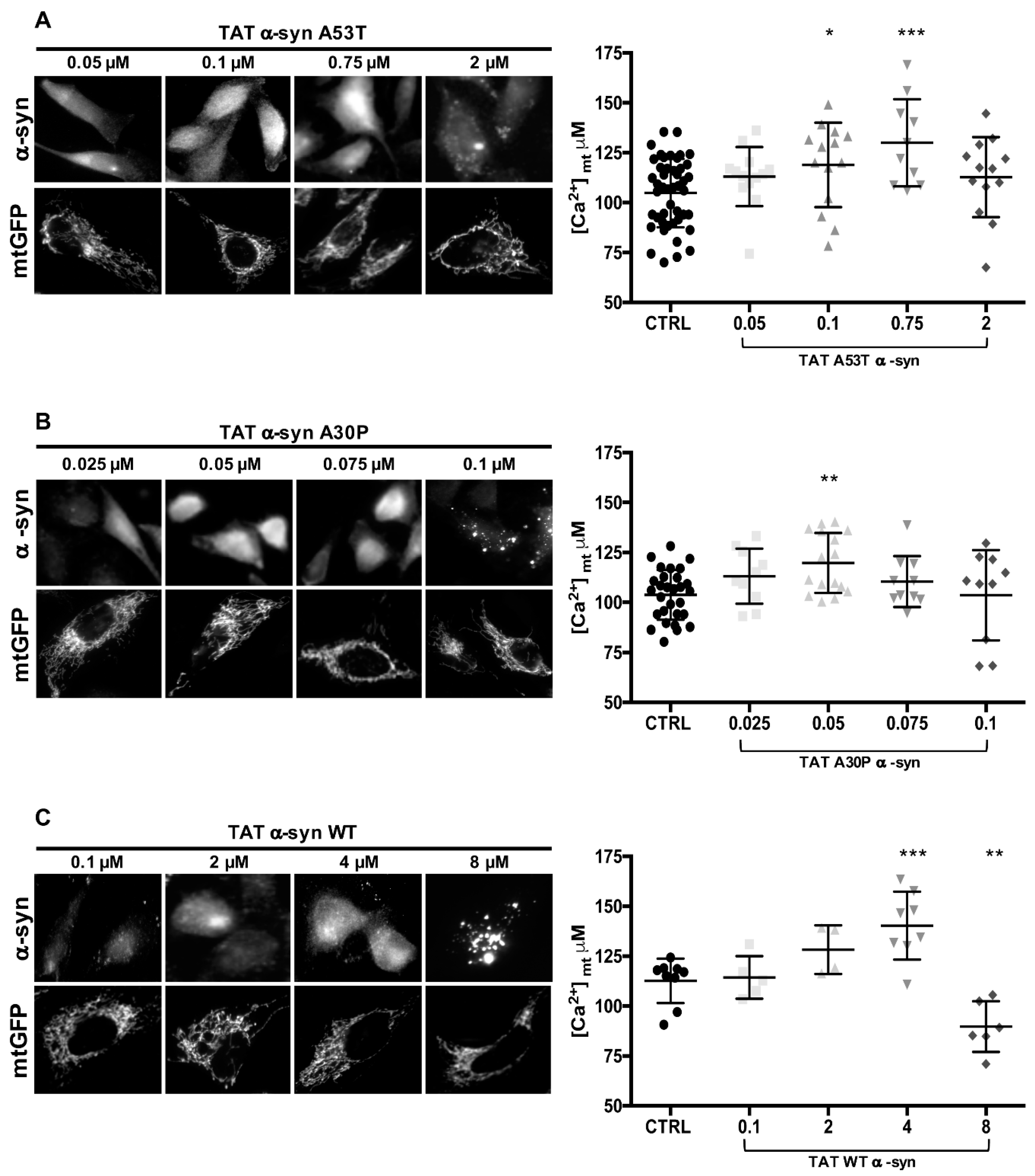
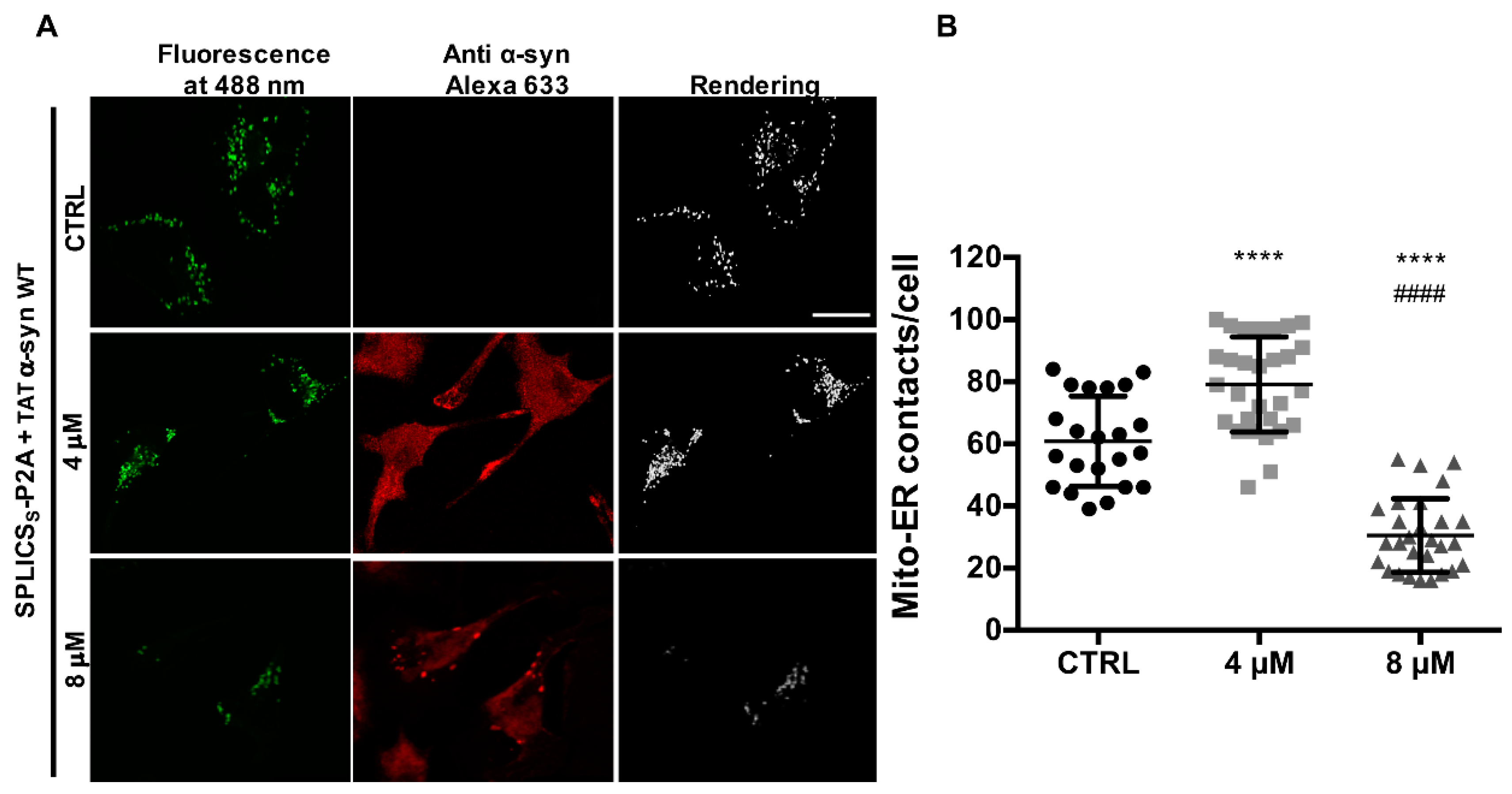
3.4. TAT-Mediated Delivery of A53T and A30P α-Synuclein Mutants Affects α-Syn Intracellular Distribution and Its Modulation of Mitochondrial Ca2+ Transients in a Dose-Dependent Manner
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bayer, T.A. Proteinopathies, a core concept for understanding and ultimately treating degenerative disorders? Eur. Neuropsychopharmacol. 2015, 25, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Cairns, N.J.; Lantos, P.L.; Goedert, M. Filamentous alpha-synuclein inclusions link multiple system atrophy with parkinson’s disease and dementia with lewy bodies. Neurosci. Lett. 1998, 251, 205–208. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S.; Faucheux, B.; Agid, Y.; Mizuno, Y.; Mochizuki, H.; Tatton, W.G.; Tatton, N.; Olanow, W.C. Dopaminergic neurons degenerate by apoptosis in parkinson’s disease. Mov. Disord. 1999, 14, 383–385. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. Alpha-synuclein in filamentous inclusions of lewy bodies from parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed]
- Kahle, P.J.; Neumann, M.; Ozmen, L.; Muller, V.; Jacobsen, H.; Schindzielorz, A.; Okochi, M.; Leimer, U.; van Der Putten, H.; Probst, A.; et al. Subcellular localization of wild-type and parkinson’s disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J. Neurosci. 2000, 20, 6365–6373. [Google Scholar] [CrossRef]
- Fujioka, S.; Ogaki, K.; Tacik, P.M.; Uitti, R.J.; Ross, O.A.; Wszolek, Z.K. Update on novel familial forms of parkinson’s disease and multiple system atrophy. Parkinsonism Relat. Disord. 2014, 20 (Suppl. 1), S29–34. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes snare-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef]
- Sun, J.; Wang, L.; Bao, H.; Premi, S.; Das, U.; Chapman, E.R.; Roy, S. Functional cooperation of alpha-synuclein and vamp2 in synaptic vesicle recycling. Proc. Natl. Acad. Sci. USA 2019, 116, 11113–11115. [Google Scholar] [CrossRef]
- Atias, M.; Tevet, Y.; Sun, J.; Stavsky, A.; Tal, S.; Kahn, J.; Roy, S.; Gitler, D. Synapsins regulate alpha-synuclein functions. Proc. Natl. Acad. Sci. USA 2019, 116, 11116–11118. [Google Scholar] [CrossRef]
- Jao, C.C.; Hegde, B.G.; Chen, J.; Haworth, I.S.; Langen, R. Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. USA 2008, 105, 19666–19671. [Google Scholar] [CrossRef]
- Eliezer, D.; Kutluay, E.; Bussell, R., Jr.; Browne, G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001, 307, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Mor, D.E.; Tsika, E.; Mazzulli, J.R.; Gould, N.S.; Kim, H.; Daniels, M.J.; Doshi, S.; Gupta, P.; Grossman, J.L.; Tan, V.X.; et al. Dopamine induces soluble alpha-synuclein oligomers and nigrostriatal degeneration. Nat. Neurosci. 2017, 20, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed]
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, prefibrillar alpha-synuclein oligomers promote complex i-dependent, ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 21490–21507. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils seed the formation of lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Goedert, M. The alpha-synucleinopathies: Parkinson’s disease, dementia with lewy bodies, and multiple system atrophy. Ann. N. Y. Acad. Sci. 2000, 920, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Interaction between alpha-synuclein and tau in parkinson’s disease comment on wills et al.: Elevated tauopathy and alpha-synuclein pathology in postmortem parkinson’s disease brains with and without dementia. Exp. Neurol. 2011, 227, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Cole, N.B.; Dieuliis, D.; Leo, P.; Mitchell, D.C.; Nussbaum, R.L. Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp. Cell Res. 2008, 314, 2076–2089. [Google Scholar] [CrossRef] [PubMed]
- Shavali, S.; Brown-Borg, H.M.; Ebadi, M.; Porter, J. Mitochondrial localization of alpha-synuclein protein in alpha-synuclein overexpressing cells. Neurosci. Lett. 2008, 439, 125–128. [Google Scholar] [CrossRef] [Green Version]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex i in human dopaminergic neuronal cultures and parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef]
- Wang, X.; Becker, K.; Levine, N.; Zhang, M.; Lieberman, A.P.; Moore, D.J.; Ma, J. Pathogenic alpha-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol. Commun. 2019, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Grassi, D.; Diaz-Perez, N.; Volpicelli-Daley, L.A.; Lasmezas, C.I. Palpha-syn* mitotoxicity is linked to mapk activation and involves tau phosphorylation and aggregation at the mitochondria. Neurobiol. Dis. 2018, 124, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. Alpha-synuclein oligomers interact with atp synthase and open the permeability transition pore in parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef] [PubMed]
- Cieri, D.; Brini, M.; Cali, T. Emerging (and converging) pathways in parkinson’s disease: Keeping mitochondrial wellness. Biochem. Biophys. Res. Commun. 2017, 483, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Vicario, M.; Cieri, D.; Brini, M.; Calì, T. The close encounter between alpha-synuclein and mitochondria. Front. Neurosci. 2018, 12, 388. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 2006, 26, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Vergnes, L.; Franich, N.R.; Reue, K.; Chesselet, M.F. Region specific mitochondrial impairment in mice with widespread overexpression of alpha-synuclein. Neurobiol. Dis. 2014, 70, 204–213. [Google Scholar] [CrossRef] [Green Version]
- Ellis, C.E.; Murphy, E.J.; Mitchell, D.C.; Golovko, M.Y.; Scaglia, F.; Barcelo-Coblijn, G.C.; Nussbaum, R.L. Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking alpha-synuclein. Mol. Cell. Biol. 2005, 25, 10190–10201. [Google Scholar] [CrossRef]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rub, C.; Liu, Y.; Magrane, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. Alpha-synuclein is localized to mitochondria-associated er membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef]
- Guardia-Laguarta, C.; Area-Gomez, E.; Schon, E.A.; Przedborski, S. A new role for alpha-synuclein in parkinson’s disease: Alteration of er-mitochondrial communication. Mov. Disord. 2015, 30, 1026–1033. [Google Scholar] [CrossRef]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. Alpha-synuclein impairs macroautophagy: Implications for parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef]
- Calì, T.; Ottolini, D.; Negro, A.; Brini, M. Alpha-synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J. Biol. Chem. 2012, 287, 17914–17929. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.H.; Fuentes, F.; Vanasco, V.; Alvarez, S.; Alaimo, A.; Cassina, A.; Coluccio Leskow, F.; Velazquez, F. Alpha-synuclein mitochondrial interaction leads to irreversible translocation and complex i impairment. Arch. Biochem. Biophys. 2018, 651, 1–12. [Google Scholar] [CrossRef]
- Ding, H.; Xiong, Y.; Sun, J.; Chen, C.; Gao, J.; Xu, H. Asiatic acid prevents oxidative stress and apoptosis by inhibiting the translocation of alpha-synuclein into mitochondria. Front. Neurosci. 2018, 12, 431. [Google Scholar] [CrossRef]
- Ma, K.L.; Song, L.K.; Yuan, Y.H.; Zhang, Y.; Han, N.; Gao, K.; Chen, N.H. The nuclear accumulation of alpha-synuclein is mediated by importin alpha and promotes neurotoxicity by accelerating the cell cycle. Neuropharmacology 2014, 82, 132–142. [Google Scholar] [CrossRef]
- Kontopoulos, E.; Parvin, J.D.; Feany, M.B. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 2006, 15, 3012–3023. [Google Scholar] [CrossRef]
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; Di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 2003, 42, 8465–8471. [Google Scholar] [CrossRef]
- Parihar, M.S.; Parihar, A.; Fujita, M.; Hashimoto, M.; Ghafourifar, P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell. Mol. Life Sci. 2008, 65, 1272–1284. [Google Scholar] [CrossRef]
- Devi, L.; Anandatheerthavarada, H.K. Mitochondrial trafficking of app and alpha synuclein: Relevance to mitochondrial dysfunction in alzheimer’s and parkinson’s diseases. Biochim. Biophys. Acta 2010, 1802, 11–19. [Google Scholar] [CrossRef]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. Alpha-synuclein binds to the er-mitochondria tethering protein vapb to disrupt ca2+ homeostasis and mitochondrial atp production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef]
- Cieri, D.; Vicario, M.; Giacomello, M.; Vallese, F.; Filadi, R.; Wagner, T.; Pozzan, T.; Pizzo, P.; Scorrano, L.; Brini, M.; et al. Splics: A split green fluorescent protein-based contact site sensor for narrow and wide heterotypic organelle juxtaposition. Cell Death Differ. 2018, 25, 1131–1145. [Google Scholar] [CrossRef]
- Albani, D.; Peverelli, E.; Rametta, R.; Batelli, S.; Veschini, L.; Negro, A.; Forloni, G. Protective effect of tat-delivered alpha-synuclein: Relevance of the c-terminal domain and involvement of hsp70. FASEB J. 2004, 18, 1713–1715. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, S.R.; Li, L.H.; Park, H.J.; Park, J.H.; Lee, K.Y.; Kim, M.K.; Shin, B.A.; Choi, S.Y. High cleavage efficiency of a 2a peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS ONE 2011, 6, e18556. [Google Scholar] [CrossRef]
- Rizzuto, R.; Brini, M.; Pizzo, P.; Murgia, M.; Pozzan, T. Chimeric green fluorescent protein as a tool for visualizing subcellular organelles in living cells. Curr. Biol. 1995, 5, 635–642. [Google Scholar] [CrossRef] [Green Version]
- Brini, M.; Marsault, R.; Bastianutto, C.; Alvarez, J.; Pozzan, T.; Rizzuto, R. Transfected aequorin in the measurement of cytosolic ca2+ concentration ([ca2+]c). A critical evaluation. J. Biol. Chem. 1995, 270, 9896–9903. [Google Scholar] [CrossRef]
- Rizzuto, R.; Simpson, A.W.; Brini, M.; Pozzan, T. Rapid changes of mitochondrial ca2+ revealed by specifically targeted recombinant aequorin. Nature 1992, 358, 325–327. [Google Scholar] [CrossRef]
- Montero, M.; Brini, M.; Marsault, R.; Alvarez, J.; Sitia, R.; Pozzan, T.; Rizzuto, R. Monitoring dynamic changes in free ca2+ concentration in the endoplasmic reticulum of intact cells. EMBO J. 1995, 14, 5467–5475. [Google Scholar] [CrossRef]
- Rizzuto, R.; Brini, M.; Bastianutto, C.; Marsault, R.; Pozzan, T. Photoprotein-mediated measurement of calcium ion concentration in mitochondria of living cells. Methods Enzymol. 1995, 260, 417–428. [Google Scholar]
- Brini, M. Calcium-sensitive photoproteins. Methods 2008, 46, 160–166. [Google Scholar] [CrossRef]
- Rizzuto, R.; Brini, M.; Pozzan, T. Targeting recombinant aequorin to specific intracellular organelles. Methods Cell Biol. 1994, 40, 339–358. [Google Scholar]
- Ottolini, D.; Cali, T.; Brini, M. Methods to measure intracellular ca(2+) fluxes with organelle-targeted aequorin-based probes. Methods Enzymol. 2014, 543, 21–45. [Google Scholar]
- Barrero, M.J.; Montero, M.; Alvarez, J. Dynamics of [ca2+] in the endoplasmic reticulum and cytoplasm of intact hela cells. A comparative study. J. Biol. Chem. 1997, 272, 27694–27699. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Cieri, D.; Vicario, M.; Vallese, F.; D’Orsi, B.; Berto, P.; Grinzato, A.; Catoni, C.; De Stefani, D.; Rizzuto, R.; Brini, M.; et al. Tau localises within mitochondrial sub-compartments and its caspase cleavage affects er-mitochondria interactions and cellular ca(2+) handling. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3247–3256. [Google Scholar] [CrossRef]
- Giacomello, M.; Drago, I.; Bortolozzi, M.; Scorzeto, M.; Gianelle, A.; Pizzo, P.; Pozzan, T. Ca2+ hot spots on the mitochondrial surface are generated by ca2+ mobilization from stores, but not by activation of store-operated ca2+ channels. Mol. Cell 2010, 38, 280–290. [Google Scholar] [CrossRef]
- Csordas, G.; Varnai, P.; Golenar, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnoczky, G. Imaging interorganelle contacts and local calcium dynamics at the er-mitochondrial interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef]
- Hettiarachchi, N.T.; Parker, A.; Dallas, M.L.; Pennington, K.; Hung, C.C.; Pearson, H.A.; Boyle, J.P.; Robinson, P.; Peers, C. Alpha-synuclein modulation of ca2+ signaling in human neuroblastoma (sh-sy5y) cells. J. Neurochem. 2009, 111, 1192–1201. [Google Scholar] [CrossRef]
- Olzscha, H.; Schermann, S.M.; Woerner, A.C.; Pinkert, S.; Hecht, M.H.; Tartaglia, G.G.; Vendruscolo, M.; Hayer-Hartl, M.; Hartl, F.U.; Vabulas, R.M. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 2011, 144, 67–78. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Lewis, P.; Revesz, T.; Lees, A.; Paisan-Ruiz, C. The genetics of parkinson’s syndromes: A critical review. Curr. Opin. Genet. Dev. 2009, 19, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Schell, H.; Hasegawa, T.; Neumann, M.; Kahle, P.J. Nuclear and neuritic distribution of serine-129 phosphorylated alpha-synuclein in transgenic mice. Neuroscience 2009, 160, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Yang, R.; Guo, J.C.; Ren, H.M.; Zha, X.L.; Cheng, J.S.; Cai, D.F. Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport 2007, 18, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Narhi, L.; Wood, S.J.; Steavenson, S.; Jiang, Y.; Wu, G.M.; Anafi, D.; Kaufman, S.A.; Martin, F.; Sitney, K.; Denis, P.; et al. Both familial parkinson’s disease mutations accelerate alpha-synuclein aggregation. J. Biol. Chem. 1999, 274, 9843–9846. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T., Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S.; Lansbury, P.T., Jr. Is there a cause-and-effect relationship between alpha-synuclein fibrillization and parkinson’s disease? Nat. Cell Biol. 2000, 2, E115–E119. [Google Scholar] [CrossRef]
- Gómez-Suaga, P.; Pérez-Nievas, B.G.; Glennon, E.B.; Lau, D.H.W.; Paillusson, S.; Mórotz, G.M.; Calì, T.; Pizzo, P.; Noble, W.; Miller, C.C.J. The vapb-ptpip51 endoplasmic reticulum-mitochondria tethering proteins are present in neuronal synapses and regulate synaptic activity. Acta Neuropathol. Commun. 2019, 7, 35. [Google Scholar] [CrossRef]
- Yeshaw, W.M.; van der Zwaag, M.; Pinto, F.; Lahaye, L.L.; Faber, A.I.; Gomez-Sanchez, R.; Dolga, A.M.; Poland, C.; Monaco, A.P.; van IJzendoorn, S.C.; et al. Human vps13a is associated with multiple organelles and influences mitochondrial morphology and lipid droplet motility. eLife 2019, 8, e43561. [Google Scholar] [CrossRef]
- Filadi, R.; Leal, N.S.; Schreiner, B.; Rossi, A.; Dentoni, G.; Pinho, C.M.; Wiehager, B.; Cieri, D.; Calì, T.; Pizzo, P.; et al. Tom70 sustains cell bioenergetics by promoting ip3r3-mediated er to mitochondria ca 2+ transfer. Curr. Biol. 2018, 28, 369–382. [Google Scholar] [CrossRef]
- Granatiero, V.; Giorgio, V.; Cali, T.; Patron, M.; Brini, M.; Bernardi, P.; Tiranti, V.; Zeviani, M.; Pallafacchina, G.; De Stefani, D.; et al. Reduced mitochondrial ca transients stimulate autophagy in human fibroblasts carrying the 13514a>g mutation of the nd5 subunit of nadh dehydrogenase. Cell Death Differ. 2016, 23, 231. [Google Scholar] [CrossRef] [PubMed]
- Doghman-Bouguerra, M.; Granatiero, V.; Sbiera, S.; Sbiera, I.; Lacas-Gervais, S.; Brau, F.; Fassnacht, M.; Rizzuto, R.; Lalli, E. Fate1 antagonizes calcium- and drug-induced apoptosis by uncoupling er and mitochondria. EMBO Rep. 2016, 17, 1264–1280. [Google Scholar] [CrossRef] [PubMed]
- Serpell, L.C.; Berriman, J.; Jakes, R.; Goedert, M.; Crowther, R.A. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc. Natl. Acad. Sci. USA 2000, 97, 4897–4902. [Google Scholar] [CrossRef]
- Li, J.; Uversky, V.N.; Fink, A.L. Effect of familial parkinson’s disease point mutations a30p and a53t on the structural properties, aggregation, and fibrillation of human alpha-synuclein. Biochemistry 2001, 40, 11604–11613. [Google Scholar] [CrossRef]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset parkinson disease. Nat. Med. 1998, 4, 1318–1320. [Google Scholar] [CrossRef]
- Lazaro, D.F.; Rodrigues, E.F.; Langohr, R.; Shahpasandzadeh, H.; Ribeiro, T.; Guerreiro, P.; Gerhardt, E.; Krohnert, K.; Klucken, J.; Pereira, M.D.; et al. Systematic comparison of the effects of alpha-synuclein mutations on its oligomerization and aggregation. PLoS Genet. 2014, 10, e1004741. [Google Scholar] [CrossRef] [PubMed]
- Pandey, N.; Schmidt, R.E.; Galvin, J.E. The alpha-synuclein mutation e46k promotes aggregation in cultured cells. Exp. Neurol. 2006, 197, 515–520. [Google Scholar] [CrossRef]
- Tosatto, L.; Horrocks, M.H.; Dear, A.J.; Knowles, T.P.; Dalla Serra, M.; Cremades, N.; Dobson, C.M.; Klenerman, D. Single-molecule fret studies on alpha-synuclein oligomerization of parkinson’s disease genetically related mutants. Sci. Rep. 2015, 5, 16696. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calì, T.; Ottolini, D.; Vicario, M.; Catoni, C.; Vallese, F.; Cieri, D.; Barazzuol, L.; Brini, M. splitGFP Technology Reveals Dose-Dependent ER-Mitochondria Interface Modulation by α-Synuclein A53T and A30P Mutants. Cells 2019, 8, 1072. https://doi.org/10.3390/cells8091072
Calì T, Ottolini D, Vicario M, Catoni C, Vallese F, Cieri D, Barazzuol L, Brini M. splitGFP Technology Reveals Dose-Dependent ER-Mitochondria Interface Modulation by α-Synuclein A53T and A30P Mutants. Cells. 2019; 8(9):1072. https://doi.org/10.3390/cells8091072
Chicago/Turabian StyleCalì, Tito, Denis Ottolini, Mattia Vicario, Cristina Catoni, Francesca Vallese, Domenico Cieri, Lucia Barazzuol, and Marisa Brini. 2019. "splitGFP Technology Reveals Dose-Dependent ER-Mitochondria Interface Modulation by α-Synuclein A53T and A30P Mutants" Cells 8, no. 9: 1072. https://doi.org/10.3390/cells8091072
APA StyleCalì, T., Ottolini, D., Vicario, M., Catoni, C., Vallese, F., Cieri, D., Barazzuol, L., & Brini, M. (2019). splitGFP Technology Reveals Dose-Dependent ER-Mitochondria Interface Modulation by α-Synuclein A53T and A30P Mutants. Cells, 8(9), 1072. https://doi.org/10.3390/cells8091072