TGFβ/BMP Signaling Pathway in Cartilage Homeostasis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Transforming Growth Factor Β (TGFβ) Family and Its Signaling
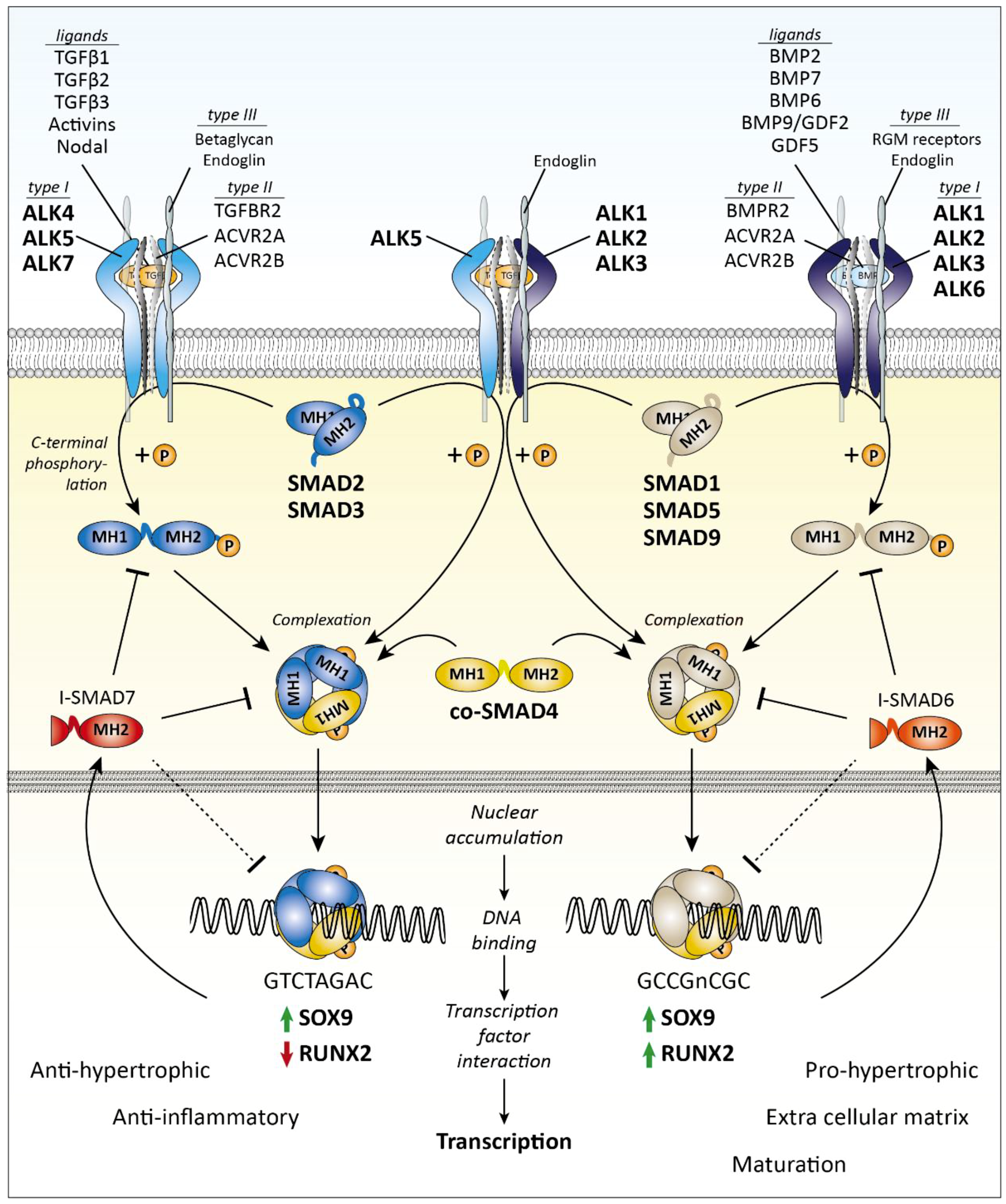
2.1. R-SMAD-Dependent Signaling
2.2. SMAD-Dependent Signaling in Chondrocytes and Cartilage Biology
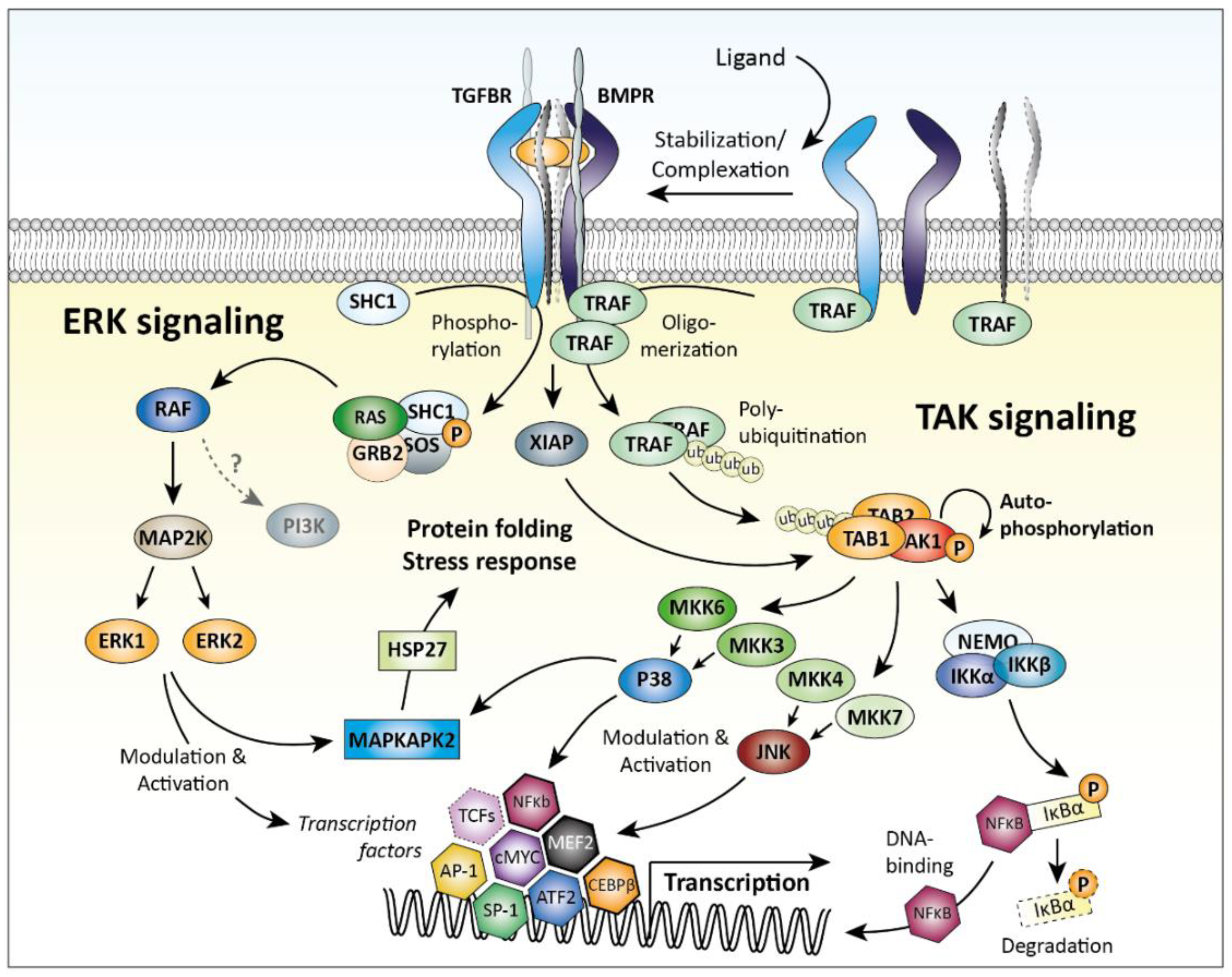
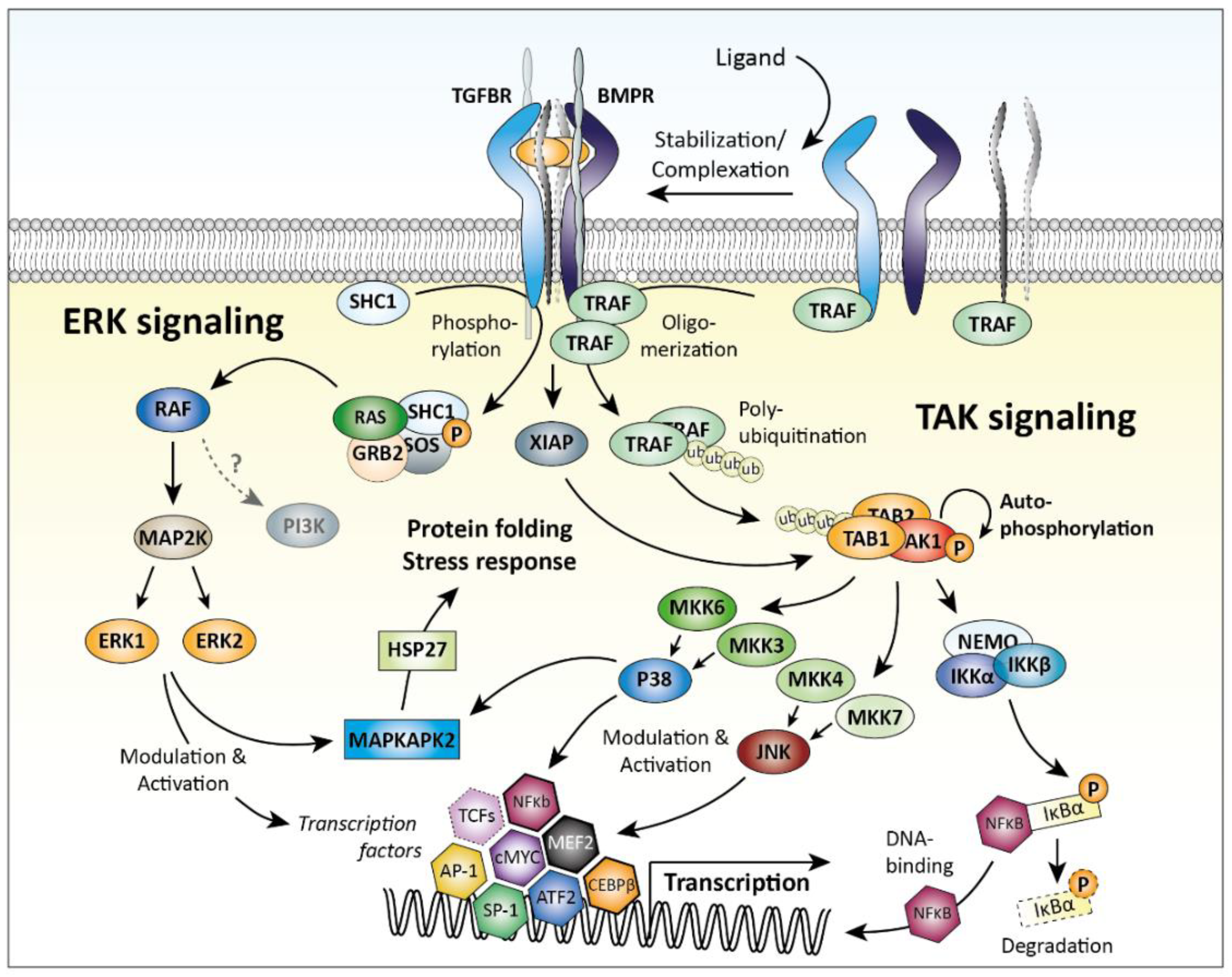
2.3. SMAD-Independent Signaling
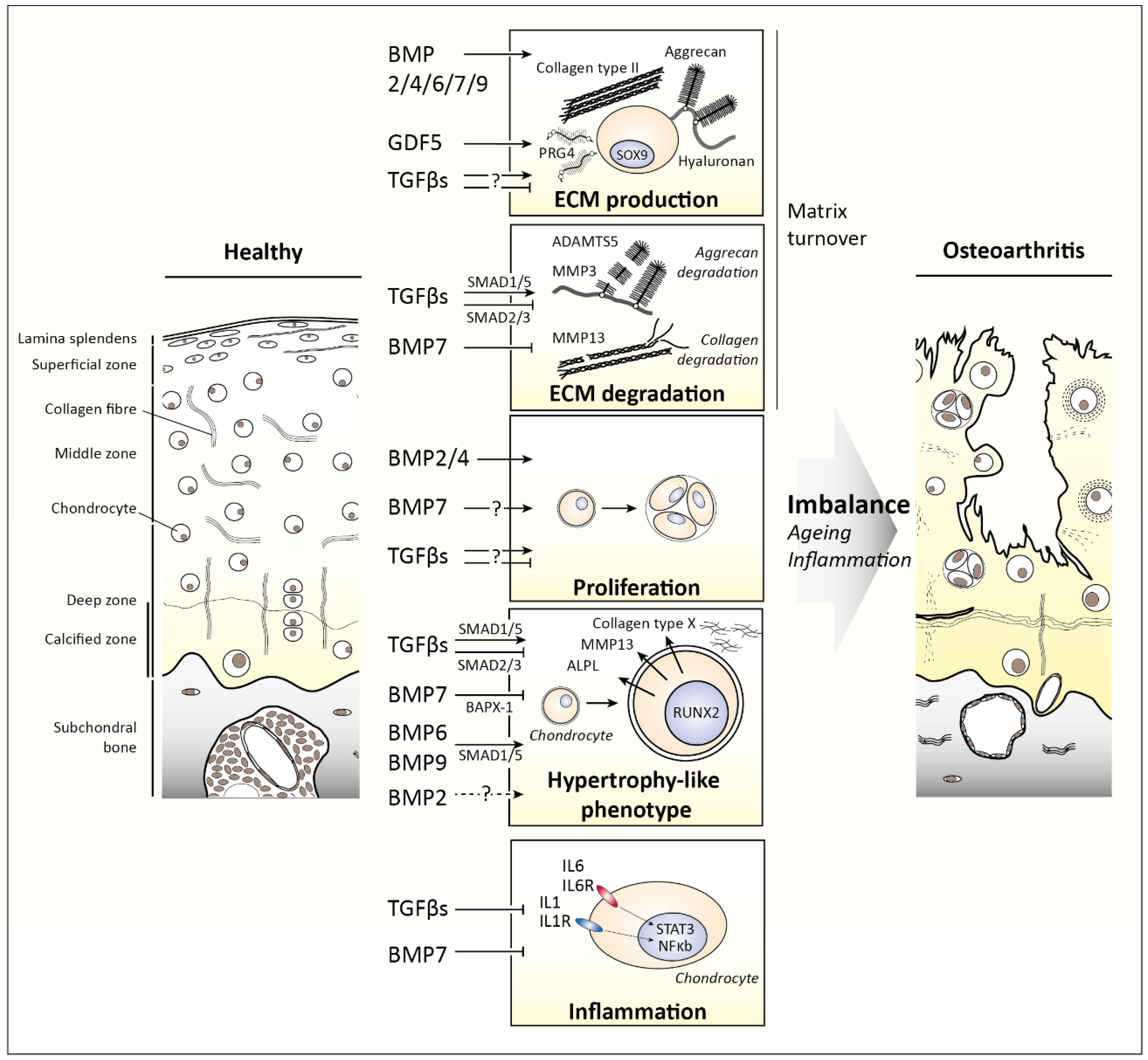
3. TGFβ Family Members in Cartilage
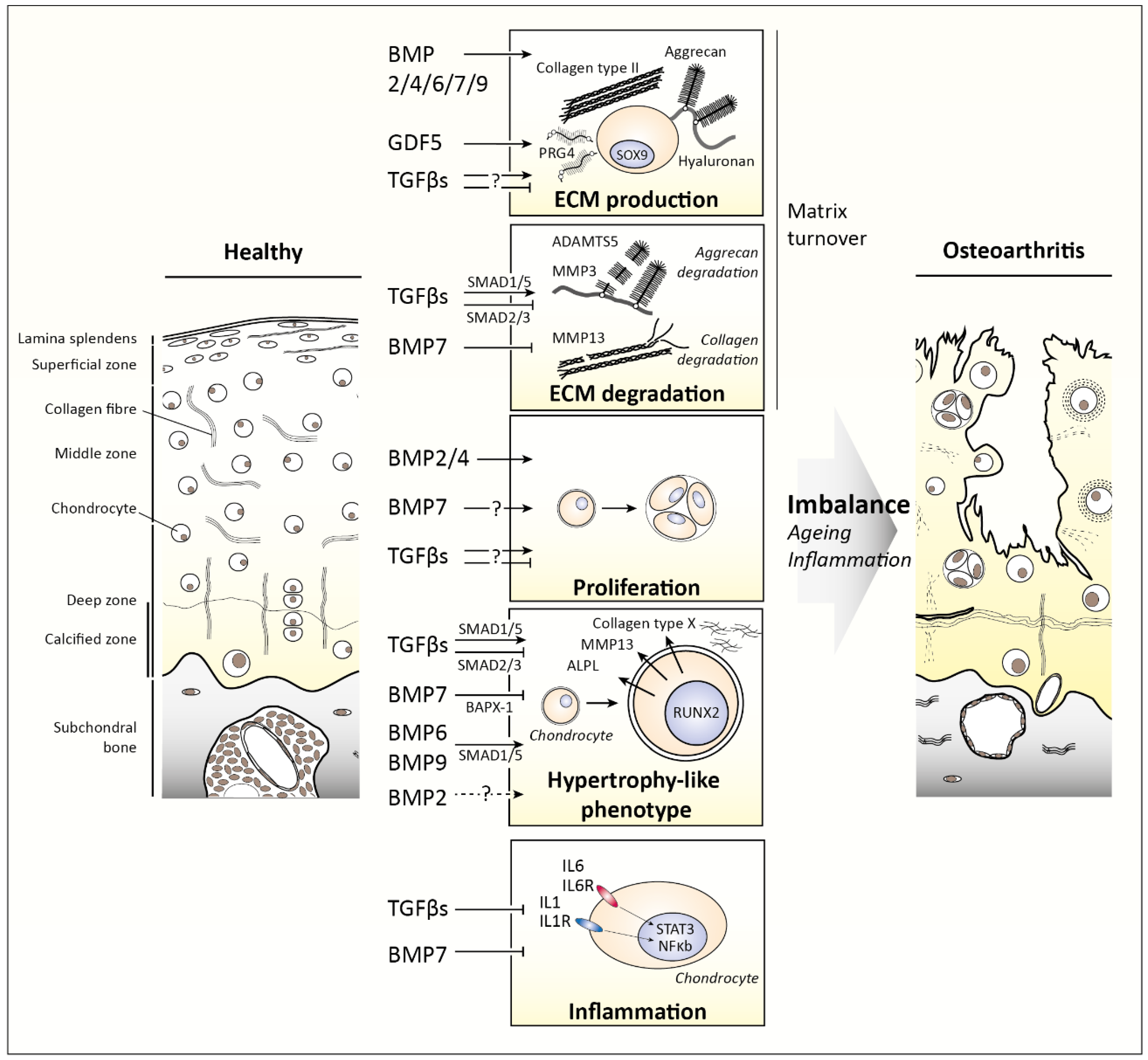
3.1. TGFβ1, TGFβ2, and TGFβ3
3.2. Bone Morphogenetic Proteins (BMPs)
4. Changes in TGFβ Family Signaling as Cause for Disease
4.1. Age-Related Changes in TGFβ Signaling
4.2. Joint Loading-Related Changes in TGFβ Signaling
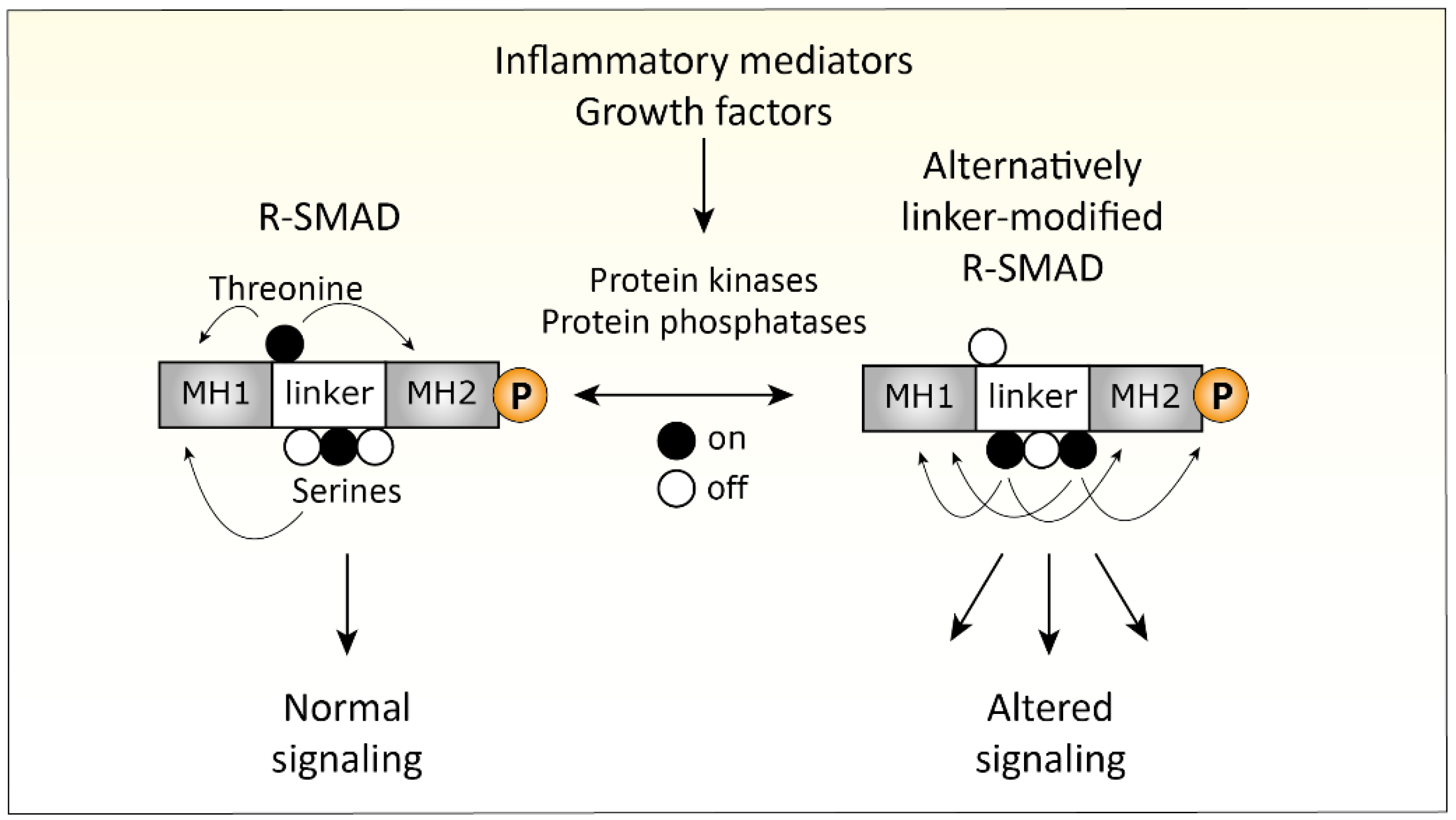
4.3. Inflammation-Related Changes in TGFβ Signaling
5. Future Perspectives
Funding
Conflicts of Interest
References
- Sophia Fox, A.J.; Bedi, A.; Rodeo, S.A. The basic science of articular cartilage: Structure, composition, and function. Sports Health 2009, 1, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Mescher, A.L.; Junqueira, L.C.U.A.B.H. Junqueira’s Basic Histology: Text and Atlas, 12th ed.; McGraw-Hill Medical: New York, NY, USA; London, UK, 2010. [Google Scholar]
- Pearle, A.D.; Warren, R.F.; Rodeo, S.A. Basic science of articular cartilage and osteoarthritis. Clin. Sports Med. 2005, 24, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shoulders, M.D.; Raines, R.T. Collagen structure and stability. Annu. Rev. Biochem. 2009, 78, 929–958. [Google Scholar] [CrossRef] [PubMed]
- Bruckner, P.; van der Rest, M. Structure and function of cartilage collagens. Microsc. Res. Tech. 1994, 28, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.R.; Laurent, T.C.; Laurent, U.B. Hyaluronan: Its nature, distribution, functions and turnover. J. Intern. Med. 1997, 242, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Kiani, C.; Chen, L.; Wu, Y.J.; Yee, A.J.; Yang, B.B. Structure and function of aggrecan. Cell Res. 2002, 12, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Teshima, R.; Otsuka, T.; Takasu, N.; Yamagata, N.; Yamamoto, K. Structure of the most superficial layer of articular cartilage. J. Bone Jt. Surg. Br. 1995, 77, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, A.K.; Blunn, G.W.; Archer, C.W.; Bentley, G. Three-dimensional collagen architecture in bovine articular cartilage. J. Bone Jt. Surg. Br. 1991, 73, 795–801. [Google Scholar] [CrossRef]
- Rhee, D.K.; Marcelino, J.; Baker, M.; Gong, Y.; Smits, P.; Lefebvre, V.; Jay, G.D.; Stewart, M.; Wang, H.; Warman, M.L.; et al. The secreted glycoprotein lubricin protects cartilage surfaces and inhibits synovial cell overgrowth. J. Clin. Investig. 2005, 115, 622–631. [Google Scholar] [CrossRef] [Green Version]
- Cowman, M.K.; Schmidt, T.A.; Raghavan, P.; Stecco, A. Viscoelastic Properties of Hyaluronan in Physiological Conditions. F1000Res 2015, 4, 622. [Google Scholar] [CrossRef] [Green Version]
- van der Kraan, P.M.; van den Berg, W.B. Chondrocyte hypertrophy and osteoarthritis: Role in initiation and progression of cartilage degeneration? Osteoarthr. Cartil. 2012, 20, 223–232. [Google Scholar] [CrossRef]
- Mueller, T.D.; Nickel, J. Promiscuity and specificity in BMP receptor activation. FEBS Lett. 2012, 586, 1846–1859. [Google Scholar] [CrossRef]
- Heldin, C.H.; Miyazono, K.; ten Dijke, P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef]
- Wang, X.F.; Lin, H.Y.; Ng-Eaton, E.; Downward, J.; Lodish, H.F.; Weinberg, R.A. Expression cloning and characterization of the TGF-beta type III receptor. Cell 1991, 67, 797–805. [Google Scholar] [CrossRef]
- Lopez-Casillas, F.; Cheifetz, S.; Doody, J.; Andres, J.L.; Lane, W.S.; Massague, J. Structure and expression of the membrane proteoglycan betaglycan, a component of the TGF-beta receptor system. Cell 1991, 67, 785–795. [Google Scholar] [CrossRef]
- Cheifetz, S.; Bellon, T.; Cales, C.; Vera, S.; Bernabeu, C.; Massague, J.; Letarte, M. Endoglin is a component of the transforming growth factor-beta receptor system in human endothelial cells. J. Biol. Chem. 1992, 267, 19027–19030. [Google Scholar]
- Siebold, C.; Yamashita, T.; Monnier, P.P.; Mueller, B.K.; Pasterkamp, R.J. RGMs: Structural Insights, Molecular Regulation, and Downstream Signaling. Trends Cell Biol. 2017, 27, 365–378. [Google Scholar] [CrossRef]
- Finnson, K.W.; Tam, B.Y.; Liu, K.; Marcoux, A.; Lepage, P.; Roy, S.; Bizet, A.A.; Philip, A. Identification of CD109 as part of the TGF-beta receptor system in human keratinocytes. FASEB J. 2006, 20, 1525–1527. [Google Scholar] [CrossRef]
- Onichtchouk, D.; Chen, Y.G.; Dosch, R.; Gawantka, V.; Delius, H.; Massague, J.; Niehrs, C. Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature 1999, 401, 480–485. [Google Scholar] [CrossRef]
- Zawel, L.; Dai, J.L.; Buckhaults, P.; Zhou, S.; Kinzler, K.W.; Vogelstein, B.; Kern, S.E. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol. Cell 1998, 1, 611–617. [Google Scholar] [CrossRef]
- Kusanagi, K.; Inoue, H.; Ishidou, Y.; Mishima, H.K.; Kawabata, M.; Miyazono, K. Characterization of a bone morphogenetic protein-responsive Smad-binding element. Mol. Biol. Cell 2000, 11, 555–565. [Google Scholar] [CrossRef]
- Gaarenstroom, T.; Hill, C.S. TGF-beta signaling to chromatin: How Smads regulate transcription during self-renewal and differentiation. Semin. Cell Dev. Biol. 2014, 32, 107–118. [Google Scholar] [CrossRef]
- Furumatsu, T.; Tsuda, M.; Taniguchi, N.; Tajima, Y.; Asahara, H. Smad3 induces chondrogenesis through the activation of SOX9 via CREB-binding protein/p300 recruitment. J. Biol. Chem. 2005, 280, 8343–8350. [Google Scholar] [CrossRef]
- Nakao, A.; Afrakhte, M.; Moren, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 1997, 389, 631–635. [Google Scholar] [CrossRef]
- Imamura, T.; Takase, M.; Nishihara, A.; Oeda, E.; Hanai, J.; Kawabata, M.; Miyazono, K. Smad6 inhibits signalling by the TGF-beta superfamily. Nature 1997, 389, 622–626. [Google Scholar] [CrossRef]
- Valdes, A.M.; Spector, T.D.; Tamm, A.; Kisand, K.; Doherty, S.A.; Dennison, E.M.; Mangino, M.; Tamm, A.; Kerna, I.; Hart, D.J.; et al. Genetic variation in the SMAD3 gene is associated with hip and knee osteoarthritis. Arthritis Rheum 2010, 62, 2347–2352. [Google Scholar] [CrossRef]
- Aref-Eshghi, E.; Zhang, Y.; Hart, D.; Valdes, A.M.; Furey, A.; Martin, G.; Sun, G.; Rahman, P.; Arden, N.; Spector, T.D.; et al. SMAD3 is associated with the total burden of radiographic osteoarthritis: The Chingford study. PLoS ONE 2014, 9, e97786. [Google Scholar] [CrossRef]
- van de Laar, I.M.; van der Linde, D.; Oei, E.H.; Bos, P.K.; Bessems, J.H.; Bierma-Zeinstra, S.M.; van Meer, B.L.; Pals, G.; Oldenburg, R.A.; Bekkers, J.A.; et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J. Med. Genet. 2012, 49, 47–57. [Google Scholar] [CrossRef]
- van de Laar, I.M.; Oldenburg, R.A.; Pals, G.; Roos-Hesselink, J.W.; de Graaf, B.M.; Verhagen, J.M.; Hoedemaekers, Y.M.; Willemsen, R.; Severijnen, L.A.; Venselaar, H.; et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 2011, 43, 121–126. [Google Scholar] [CrossRef]
- Yang, X.; Chen, L.; Xu, X.; Li, C.; Huang, C.; Deng, C.X. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol. 2001, 153, 35–46. [Google Scholar] [CrossRef]
- Chen, C.G.; Thuillier, D.; Chin, E.N.; Alliston, T. Chondrocyte-intrinsic Smad3 represses Runx2-inducible matrix metalloproteinase 13 expression to maintain articular cartilage and prevent osteoarthritis. Arthritis Rheum 2012, 64, 3278–3289. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, C.M.; Schwarz, E.M.; Reynolds, P.R.; Puzas, J.E.; Rosier, R.N.; O’Keefe, R.J. Smad2 and 3 mediate transforming growth factor-beta1-induced inhibition of chondrocyte maturation. Endocrinology 2000, 141, 4728–4735. [Google Scholar] [CrossRef]
- Ionescu, A.M.; Schwarz, E.M.; Zuscik, M.J.; Drissi, H.; Puzas, J.E.; Rosier, R.N.; O’Keefe, R.J. ATF-2 cooperates with Smad3 to mediate TGF-beta effects on chondrocyte maturation. Exp. Cell Res. 2003, 288, 198–207. [Google Scholar] [CrossRef]
- Kim, K.O.; Sampson, E.R.; Maynard, R.D.; O’Keefe, R.J.; Chen, D.; Drissi, H.; Rosier, R.N.; Hilton, M.J.; Zuscik, M.J. Ski inhibits TGF-beta/phospho-Smad3 signaling and accelerates hypertrophic differentiation in chondrocytes. J. Cell Biochem. 2012, 113, 2156–2166. [Google Scholar] [CrossRef]
- Li, T.F.; Gao, L.; Sheu, T.J.; Sampson, E.R.; Flick, L.M.; Konttinen, Y.T.; Chen, D.; Schwarz, E.M.; Zuscik, M.J.; Jonason, J.H.; et al. Aberrant hypertrophy in Smad3-deficient murine chondrocytes is rescued by restoring transforming growth factor beta-activated kinase 1/activating transcription factor 2 signaling: a potential clinical implication for osteoarthritis. Arthritis Rheum 2010, 62, 2359–2369. [Google Scholar] [CrossRef]
- Kang, J.S.; Alliston, T.; Delston, R.; Derynck, R. Repression of Runx2 function by TGF-beta through recruitment of class II histone deacetylases by Smad3. EMBO J. 2005, 24, 2543–2555. [Google Scholar] [CrossRef]
- Leboy, P.; Grasso-Knight, G.; D’Angelo, M.; Volk, S.W.; Lian, J.V.; Drissi, H.; Stein, G.S.; Adams, S.L. Smad-Runx interactions during chondrocyte maturation. J. Bone Jt. Surg. Am. 2001, 83-A Suppl 1 (Pt 1), S15–S22. [Google Scholar] [CrossRef]
- Takeda, S.; Bonnamy, J.P.; Owen, M.J.; Ducy, P.; Karsenty, G. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 2001, 15, 467–481. [Google Scholar] [CrossRef]
- Enomoto, H.; Enomoto-Iwamoto, M.; Iwamoto, M.; Nomura, S.; Himeno, M.; Kitamura, Y.; Kishimoto, T.; Komori, T. Cbfa1 is a positive regulatory factor in chondrocyte maturation. J. Biol. Chem. 2000, 275, 8695–8702. [Google Scholar] [CrossRef]
- Inada, M.; Yasui, T.; Nomura, S.; Miyake, S.; Deguchi, K.; Himeno, M.; Sato, M.; Yamagiwa, H.; Kimura, T.; Yasui, N.; et al. Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev. Dyn. 1999, 214, 279–290. [Google Scholar] [CrossRef]
- Nomura, M.; Li, E. Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature 1998, 393, 786–790. [Google Scholar] [CrossRef]
- Waldrip, W.R.; Bikoff, E.K.; Hoodless, P.A.; Wrana, J.L.; Robertson, E.J. Smad2 signaling in extraembryonic tissues determines anterior-posterior polarity of the early mouse embryo. Cell 1998, 92, 797–808. [Google Scholar] [CrossRef]
- Weinstein, M.; Yang, X.; Li, C.; Xu, X.; Gotay, J.; Deng, C.X. Failure of egg cylinder elongation and mesoderm induction in mouse embryos lacking the tumor suppressor smad2. Proc. Natl. Acad. Sci. USA 1998, 95, 9378–9383. [Google Scholar] [CrossRef]
- Vivian, J.L.; Chen, Y.; Yee, D.; Schneider, E.; Magnuson, T. An allelic series of mutations in Smad2 and Smad4 identified in a genotype-based screen of N-ethyl-N- nitrosourea-mutagenized mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA 2002, 99, 15542–15547. [Google Scholar] [CrossRef]
- Decker, R.S.; Koyama, E.; Pacifici, M. Genesis and morphogenesis of limb synovial joints and articular cartilage. Matrix Biol. 2014, 39, 5–10. [Google Scholar] [CrossRef]
- Hamamoto, T.; Beppu, H.; Okada, H.; Kawabata, M.; Kitamura, T.; Miyazono, K.; Kato, M. Compound disruption of smad2 accelerates malignant progression of intestinal tumors in apc knockout mice. Cancer Res. 2002, 62, 5955–5961. [Google Scholar]
- Wang, W.; Song, B.; Anbarchian, T.; Shirazyan, A.; Sadik, J.E.; Lyons, K.M. Smad2 and Smad3 Regulate Chondrocyte Proliferation and Differentiation in the Growth Plate. PLoS Genet. 2016, 12, e1006352. [Google Scholar] [CrossRef]
- Lechleider, R.J.; Ryan, J.L.; Garrett, L.; Eng, C.; Deng, C.; Wynshaw-Boris, A.; Roberts, A.B. Targeted mutagenesis of Smad1 reveals an essential role in chorioallantoic fusion. Dev. Biol. 2001, 240, 157–167. [Google Scholar] [CrossRef]
- Chang, H.; Huylebroeck, D.; Verschueren, K.; Guo, Q.; Matzuk, M.M.; Zwijsen, A. Smad5 knockout mice die at mid-gestation due to multiple embryonic and extraembryonic defects. Development 1999, 126, 1631–1642. [Google Scholar]
- Retting, K.N.; Song, B.; Yoon, B.S.; Lyons, K.M. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 2009, 136, 1093–1104. [Google Scholar] [CrossRef]
- Fujii, M.; Takeda, K.; Imamura, T.; Aoki, H.; Sampath, T.K.; Enomoto, S.; Kawabata, M.; Kato, M.; Ichijo, H.; Miyazono, K. Roles of bone morphogenetic protein type I receptors and Smad proteins in osteoblast and chondroblast differentiation. Mol. Biol. Cell 1999, 10, 3801–3813. [Google Scholar] [CrossRef]
- Horiki, M.; Imamura, T.; Okamoto, M.; Hayashi, M.; Murai, J.; Myoui, A.; Ochi, T.; Miyazono, K.; Yoshikawa, H.; Tsumaki, N. Smad6/Smurf1 overexpression in cartilage delays chondrocyte hypertrophy and causes dwarfism with osteopenia. J. Cell Biol. 2004, 165, 433–445. [Google Scholar] [CrossRef]
- Javed, A.; Afzal, F.; Bae, J.S.; Gutierrez, S.; Zaidi, K.; Pratap, J.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B. Specific residues of RUNX2 are obligatory for formation of BMP2-induced RUNX2-SMAD complex to promote osteoblast differentiation. Cells Tissues Organs 2009, 189, 133–137. [Google Scholar] [CrossRef]
- Javed, A.; Bae, J.S.; Afzal, F.; Gutierrez, S.; Pratap, J.; Zaidi, S.K.; Lou, Y.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; et al. Structural coupling of Smad and Runx2 for execution of the BMP2 osteogenic signal. J. Biol. Chem. 2008, 283, 8412–8422. [Google Scholar] [CrossRef]
- Phimphilai, M.; Zhao, Z.; Boules, H.; Roca, H.; Franceschi, R.T. BMP signaling is required for RUNX2-dependent induction of the osteoblast phenotype. J. Bone Miner. Res. 2006, 21, 637–646. [Google Scholar] [CrossRef]
- Bae, J.S.; Gutierrez, S.; Narla, R.; Pratap, J.; Devados, R.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B.; Javed, A. Reconstitution of Runx2/Cbfa1-null cells identifies a requirement for BMP2 signaling through a Runx2 functional domain during osteoblast differentiation. J. Cell Biochem. 2007, 100, 434–449. [Google Scholar] [CrossRef]
- Kielty, C.M.; Kwan, A.P.; Holmes, D.F.; Schor, S.L.; Grant, M.E. Type X collagen, a product of hypertrophic chondrocytes. Biochem. J. 1985, 227, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef]
- Landstrom, M. The TAK1-TRAF6 signalling pathway. Int. J. Biochem. Cell Biol. 2010, 42, 585–589. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Nagai, S.; Ninomiya-Tsuji, J.; Nishita, M.; Tamai, K.; Irie, K.; Ueno, N.; Nishida, E.; Shibuya, H.; Matsumoto, K. XIAP, a cellular member of the inhibitor of apoptosis protein family, links the receptors to TAB1-TAK1 in the BMP signaling pathway. EMBO J. 1999, 18, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Galban, S.; Duckett, C.S. XIAP as a ubiquitin ligase in cellular signaling. Cell Death Differ. 2010, 17, 54–60. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Watanabe, H.; de Caestecker, M.P.; Yamada, Y. Transcriptional cross-talk between Smad, ERK1/2, and p38 mitogen-activated protein kinase pathways regulates transforming growth factor-beta-induced aggrecan gene expression in chondrogenic ATDC5 cells. J. Biol. Chem. 2001, 276, 14466–14473. [Google Scholar] [CrossRef]
- Zhu, Y.; Tao, H.; Jin, C.; Liu, Y.; Lu, X.; Hu, X.; Wang, X. Transforming growth factor-beta1 induces type II collagen and aggrecan expression via activation of extracellular signal-regulated kinase 1/2 and Smad2/3 signaling pathways. Mol. Med. Rep. 2015, 12, 5573–5579. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Tsukazaki, T.; Hirota, Y.; Yonekura, A.; Osaki, M.; Shindo, H.; Yamashita, S. Dexamethasone inhibition of TGF beta-induced cell growth and type II collagen mRNA expression through ERK-integrated AP-1 activity in cultured rat articular chondrocytes. Osteoarthr. Cartil. 2000, 8, 378–385. [Google Scholar] [CrossRef]
- Qureshi, H.Y.; Sylvester, J.; El Mabrouk, M.; Zafarullah, M. TGF-beta-induced expression of tissue inhibitor of metalloproteinases-3 gene in chondrocytes is mediated by extracellular signal-regulated kinase pathway and Sp1 transcription factor. J. Cell Physiol. 2005, 203, 345–352. [Google Scholar] [CrossRef]
- Tuli, R.; Tuli, S.; Nandi, S.; Huang, X.; Manner, P.A.; Hozack, W.J.; Danielson, K.G.; Hall, D.J.; Tuan, R.S. Transforming growth factor-beta-mediated chondrogenesis of human mesenchymal progenitor cells involves N-cadherin and mitogen-activated protein kinase and Wnt signaling cross-talk. J. Biol. Chem. 2003, 278, 41227–41236. [Google Scholar] [CrossRef]
- Li, J.; Zhao, Z.; Liu, J.; Huang, N.; Long, D.; Wang, J.; Li, X.; Liu, Y. MEK/ERK and p38 MAPK regulate chondrogenesis of rat bone marrow mesenchymal stem cells through delicate interaction with TGF-beta1/Smads pathway. Cell Prolif. 2010, 43, 333–343. [Google Scholar] [CrossRef]
- Bobick, B.E.; Kulyk, W.M. The MEK-ERK signaling pathway is a negative regulator of cartilage-specific gene expression in embryonic limb mesenchyme. J. Biol. Chem. 2004, 279, 4588–4595. [Google Scholar] [CrossRef]
- Qiao, B.; Padilla, S.R.; Benya, P.D. Transforming growth factor (TGF)-beta-activated kinase 1 mimics and mediates TGF-beta-induced stimulation of type II collagen synthesis in chondrocytes independent of Col2a1 transcription and Smad3 signaling. J. Biol. Chem. 2005, 280, 17562–17571. [Google Scholar] [CrossRef]
- Gunnell, L.M.; Jonason, J.H.; Loiselle, A.E.; Kohn, A.; Schwarz, E.M.; Hilton, M.J.; O’Keefe, R.J. TAK1 regulates cartilage and joint development via the MAPK and BMP signaling pathways. J. Bone Miner. Res. 2010, 25, 1784–1797. [Google Scholar] [CrossRef] [Green Version]
- Shim, J.H.; Greenblatt, M.B.; Xie, M.; Schneider, M.D.; Zou, W.; Zhai, B.; Gygi, S.; Glimcher, L.H. TAK1 is an essential regulator of BMP signalling in cartilage. EMBO J. 2009, 28, 2028–2041. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Sheu, T.J.; Dong, Y.; Hoak, D.M.; Zuscik, M.J.; Schwarz, E.M.; Hilton, M.J.; O’Keefe, R.J.; Jonason, J.H. TAK1 regulates SOX9 expression in chondrocytes and is essential for postnatal development of the growth plate and articular cartilages. J. Cell Sci. 2013, 126, 5704–5713. [Google Scholar] [CrossRef] [Green Version]
- Tew, S.R.; Hardingham, T.E. Regulation of SOX9 mRNA in human articular chondrocytes involving p38 MAPK activation and mRNA stabilization. J. Biol. Chem. 2006, 281, 39471–39479. [Google Scholar] [CrossRef]
- Rokutanda, S.; Fujita, T.; Kanatani, N.; Yoshida, C.A.; Komori, H.; Liu, W.; Mizuno, A.; Komori, T. Akt regulates skeletal development through GSK3, mTOR, and FoxOs. Dev. Biol. 2009, 328, 78–93. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.; Zhang, Z.; Jin, D.; Cai, C.; Jia, C.; Liu, W.; Wang, T.; Li, S.; Zhang, H.; Huang, B.; et al. mTORC1 regulates PTHrP to coordinate chondrocyte growth, proliferation and differentiation. Nat. Commun. 2016, 7, 11151. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Vasheghani, F.; Li, Y.-h.; Blati, M.; Simeone, K.; Fahmi, H.; Lussier, B.; Roughley, P.; Lagares, D.; Pelletier, J.-P.; et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann. Rheum. Dis. 2015, 74, 1432–1440. [Google Scholar] [CrossRef]
- Takayama, K.; Kawakami, Y.; Kobayashi, M.; Greco, N.; Cummins, J.H.; Matsushita, T.; Kuroda, R.; Kurosaka, M.; Fu, F.H.; Huard, J. Local intra-articular injection of rapamycin delays articular cartilage degeneration in a murine model of osteoarthritis. Arthritis Res. Ther. 2014, 16, 482. [Google Scholar] [CrossRef]
- Carames, B.; Hasegawa, A.; Taniguchi, N.; Miyaki, S.; Blanco, F.J.; Lotz, M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann. Rheum. Dis. 2012, 71, 575–581. [Google Scholar] [CrossRef]
- Woods, A.; Wang, G.; Beier, F. RhoA/ROCK signaling regulates Sox9 expression and actin organization during chondrogenesis. J. Biol. Chem. 2005, 280, 11626–11634. [Google Scholar] [CrossRef]
- Woods, A.; Beier, F. RhoA/ROCK signaling regulates chondrogenesis in a context-dependent manner. J. Biol. Chem. 2006, 281, 13134–13140. [Google Scholar] [CrossRef]
- Haudenschild, D.R.; Chen, J.; Pang, N.; Lotz, M.K.; D’Lima, D.D. Rho kinase-dependent activation of SOX9 in chondrocytes. Arthritis Rheum 2010, 62, 191–200. [Google Scholar] [CrossRef]
- Seyedin, S.M.; Thomas, T.C.; Thompson, A.Y.; Rosen, D.M.; Piez, K.A. Purification and characterization of two cartilage-inducing factors from bovine demineralized bone. Proc. Natl. Acad. Sci. USA 1985, 82, 2267–2271. [Google Scholar] [CrossRef]
- Seyedin, S.M.; Thompson, A.Y.; Bentz, H.; Rosen, D.M.; McPherson, J.M.; Conti, A.; Siegel, N.R.; Galluppi, G.R.; Piez, K.A. Cartilage-inducing factor-A. Apparent identity to transforming growth factor-beta. J. Biol. Chem. 1986, 261, 5693–5695. [Google Scholar]
- Segarini, P.R.; Roberts, A.B.; Rosen, D.M.; Seyedin, S.M. Membrane binding characteristics of two forms of transforming growth factor-beta. J. Biol. Chem. 1987, 262, 14655–14662. [Google Scholar]
- Ogawa, Y.; Schmidt, D.K.; Dasch, J.R.; Chang, R.J.; Glaser, C.B. Purification and characterization of transforming growth factor-beta 2.3 and -beta 1.2 heterodimers from bovine bone. J. Biol. Chem. 1992, 267, 2325–2328. [Google Scholar]
- Sengle, G.; Ono, R.N.; Sasaki, T.; Sakai, L.Y. Prodomains of transforming growth factor beta (TGFbeta) superfamily members specify different functions: extracellular matrix interactions and growth factor bioavailability. J. Biol. Chem. 2011, 286, 5087–5099. [Google Scholar] [CrossRef]
- Blaney Davidson, E.N.; Scharstuhl, A.; Vitters, E.L.; van der Kraan, P.M.; van den Berg, W.B. Reduced transforming growth factor-beta signaling in cartilage of old mice: Role in impaired repair capacity. Arthritis Res. Ther. 2005, 7, R1338–R1347. [Google Scholar] [CrossRef]
- Villiger, P.M.; Lotz, M. Differential expression of TGF beta isoforms by human articular chondrocytes in response to growth factors. J. Cell Physiol. 1992, 151, 318–325. [Google Scholar] [CrossRef]
- Pombo-Suarez, M.; Castano-Oreja, M.T.; Calaza, M.; Gomez-Reino, J.; Gonzalez, A. Differential upregulation of the three transforming growth factor beta isoforms in human osteoarthritic cartilage. Ann. Rheum. Dis. 2009, 68, 568–571. [Google Scholar] [CrossRef]
- Morales, T.I.; Joyce, M.E.; Sobel, M.E.; Danielpour, D.; Roberts, A.B. Transforming growth factor-beta in calf articular cartilage organ cultures: Synthesis and distribution. Arch. Biochem. Biophys. 1991, 288, 397–405. [Google Scholar] [CrossRef]
- Albro, M.B.; Nims, R.J.; Cigan, A.D.; Yeroushalmi, K.J.; Shim, J.J.; Hung, C.T.; Ateshian, G.A. Dynamic mechanical compression of devitalized articular cartilage does not activate latent TGF-beta. J. Biomech. 2013, 46, 1433–1439. [Google Scholar] [CrossRef]
- Maeda, S.; Dean, D.D.; Gay, I.; Schwartz, Z.; Boyan, B.D. Activation of latent transforming growth factor beta1 by stromelysin 1 in extracts of growth plate chondrocyte-derived matrix vesicles. J. Bone Miner. Res. 2001, 16, 1281–1290. [Google Scholar] [CrossRef]
- Albro, M.B.; Cigan, A.D.; Nims, R.J.; Yeroushalmi, K.J.; Oungoulian, S.R.; Hung, C.T.; Ateshian, G.A. Shearing of synovial fluid activates latent TGF-beta. Osteoarthr. Cartil. 2012, 20, 1374–1382. [Google Scholar] [CrossRef]
- Jobling, M.F.; Mott, J.D.; Finnegan, M.T.; Jurukovski, V.; Erickson, A.C.; Walian, P.J.; Taylor, S.E.; Ledbetter, S.; Lawrence, C.M.; Rifkin, D.B.; et al. Isoform-specific activation of latent transforming growth factor beta (LTGF-beta) by reactive oxygen species. Radiat. Res. 2006, 166, 839–848. [Google Scholar] [CrossRef]
- Madej, W.; van Caam, A.; Blaney Davidson, E.N.; van der Kraan, P.M.; Buma, P. Physiological and excessive mechanical compression of articular cartilage activates Smad2/3P signaling. Osteoarthr. Cartil. 2014, 22, 1018–1025. [Google Scholar] [CrossRef] [Green Version]
- Finnson, K.W.; Parker, W.L.; ten Dijke, P.; Thorikay, M.; Philip, A. ALK1 opposes ALK5/Smad3 signaling and expression of extracellular matrix components in human chondrocytes. J. Bone Miner. Res. 2008, 23, 896–906. [Google Scholar] [CrossRef]
- Blaney Davidson, E.N.; Remst, D.F.; Vitters, E.L.; van Beuningen, H.M.; Blom, A.B.; Goumans, M.J.; van den Berg, W.B.; van der Kraan, P.M. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J. Immunol. 2009, 182, 7937–7945. [Google Scholar] [CrossRef]
- van Beuningen, H.M.; van der Kraan, P.M.; Arntz, O.J.; van den Berg, W.B. Transforming growth factor-beta 1 stimulates articular chondrocyte proteoglycan synthesis and induces osteophyte formation in the murine knee joint. Lab. Investig. 1994, 71, 279–290. [Google Scholar]
- van Beuningen, H.M.; Glansbeek, H.L.; van der Kraan, P.M.; van den Berg, W.B. Differential effects of local application of BMP-2 or TGF-beta 1 on both articular cartilage composition and osteophyte formation. Osteoarthr. Cartil. 1998, 6, 306–317. [Google Scholar] [CrossRef]
- Morales, T.I.; Roberts, A.B. Transforming growth factor beta regulates the metabolism of proteoglycans in bovine cartilage organ cultures. J. Biol. Chem. 1988, 263, 12828–12831. [Google Scholar]
- Malemud, C.J.; Killeen, W.; Hering, T.M.; Purchio, A.F. Enhanced sulfated-proteoglycan core protein synthesis by incubation of rabbit chondrocytes with recombinant transforming growth factor-beta 1. J. Cell Physiol. 1991, 149, 152–159. [Google Scholar] [CrossRef]
- van der Kraan, P.; Vitters, E.; van den Berg, W. Differential effect of transforming growth factor beta on freshly isolated and cultured articular chondrocytes. J. Rheumatol. 1992, 19, 140–145. [Google Scholar]
- Redini, F.; Galera, P.; Mauviel, A.; Loyau, G.; Pujol, J.P. Transforming growth factor beta stimulates collagen and glycosaminoglycan biosynthesis in cultured rabbit articular chondrocytes. FEBS Lett. 1988, 234, 172–176. [Google Scholar] [CrossRef]
- Galera, P.; Vivien, D.; Pronost, S.; Bonaventure, J.; Redini, F.; Loyau, G.; Pujol, J.P. Transforming growth factor-beta 1 (TGF-beta 1) up-regulation of collagen type II in primary cultures of rabbit articular chondrocytes (RAC) involves increased mRNA levels without affecting mRNA stability and procollagen processing. J. Cell Physiol. 1992, 153, 596–606. [Google Scholar] [CrossRef]
- Recklies, A.D.; Baillargeon, L.; White, C. Regulation of cartilage oligomeric matrix protein synthesis in human synovial cells and articular chondrocytes. Arthritis Rheum 1998, 41, 997–1006. [Google Scholar] [CrossRef]
- Motaung, S.C.; Di Cesare, P.E.; Reddi, A.H. Differential response of cartilage oligomeric matrix protein (COMP) to morphogens of bone morphogenetic protein/transforming growth factor-beta family in the surface, middle and deep zones of articular cartilage. J. Tissue Eng. Regen. Med. 2011, 5, e87–e96. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Pillarisetti, J.; Sharma, B.; Murdoch, A.D.; Danielson, K.G.; Uitto, J.; Mauviel, A. Structural and functional characterization of the human perlecan gene promoter. Transcriptional activation by transforming growth factor-beta via a nuclear factor 1-binding element. J. Biol. Chem. 1997, 272, 5219–5228. [Google Scholar] [CrossRef]
- Burton-Wurster, N.; Lust, G. Fibronectin and proteoglycan synthesis in long term cultures of cartilage explants in Ham’s F12 supplemented with insulin and calcium: effects of the addition of TGF-beta. Arch. Biochem. Biophys. 1990, 283, 27–33. [Google Scholar] [CrossRef]
- Morales, T.I. Transforming growth factor-beta 1 stimulates synthesis of proteoglycan aggregates in calf articular cartilage organ cultures. Arch. Biochem. Biophys. 1991, 286, 99–106. [Google Scholar] [CrossRef]
- Niikura, T.; Reddi, A.H. Differential regulation of lubricin/superficial zone protein by transforming growth factor beta/bone morphogenetic protein superfamily members in articular chondrocytes and synoviocytes. Arthritis Rheum 2007, 56, 2312–2321. [Google Scholar] [CrossRef]
- van der Kraan, P.M.; Vitters, E.L.; van den Berg, W.B. Inhibition of proteoglycan synthesis by transforming growth factor beta in anatomically intact articular cartilage of murine patellae. Ann. Rheum. Dis. 1992, 51, 643–647. [Google Scholar] [CrossRef]
- Glansbeek, H.L.; van der Kraan, P.M.; Vitters, E.L.; van den Berg, W.B. Correlation of the size of type II transforming growth factor beta (TGF-beta) receptor with TGF-beta responses of isolated bovine articular chondrocytes. Ann. Rheum. Dis. 1993, 52, 812–816. [Google Scholar] [CrossRef]
- Chadjichristos, C.; Ghayor, C.; Herrouin, J.F.; Ala-Kokko, L.; Suske, G.; Pujol, J.P.; Galera, P. Down-regulation of human type II collagen gene expression by transforming growth factor-beta 1 (TGF-beta 1) in articular chondrocytes involves SP3/SP1 ratio. J. Biol. Chem. 2002, 277, 43903–43917. [Google Scholar] [CrossRef]
- O’Keefe, R.J.; Puzas, J.E.; Brand, J.S.; Rosier, R.N. Effects of transforming growth factor-beta on matrix synthesis by chick growth plate chondrocytes. Endocrinology 1988, 122, 2953–2961. [Google Scholar] [CrossRef]
- Vivien, D.; Galera, P.; Lebrun, E.; Loyau, G.; Pujol, J.P. Differential effects of transforming growth factor-beta and epidermal growth factor on the cell cycle of cultured rabbit articular chondrocytes. J. Cell Physiol. 1990, 143, 534–545. [Google Scholar] [CrossRef]
- van Beuningen, H.M.; Glansbeek, H.L.; van der Kraan, P.M.; van den Berg, W.B. Osteoarthritis-like changes in the murine knee joint resulting from intra-articular transforming growth factor-beta injections. Osteoarthr. Cartil. 2000, 8, 25–33. [Google Scholar] [CrossRef]
- van Beuningen, H.M.; van der Kraan, P.M.; Arntz, O.J.; van den Berg, W.B. Protection from interleukin 1 induced destruction of articular cartilage by transforming growth factor beta: studies in anatomically intact cartilage in vitro and in vivo. Ann. Rheum. Dis. 1993, 52, 185–191. [Google Scholar] [CrossRef]
- van Beuningen, H.M.; van der Kraan, P.M.; Arntz, O.J.; van den Berg, W.B. In vivo protection against interleukin-1-induced articular cartilage damage by transforming growth factor-beta 1: age-related differences. Ann. Rheum. Dis. 1994, 53, 593–600. [Google Scholar] [CrossRef]
- Takahashi, N.; Rieneck, K.; van der Kraan, P.M.; van Beuningen, H.M.; Vitters, E.L.; Bendtzen, K.; van den Berg, W.B. Elucidation of IL-1/TGF-beta interactions in mouse chondrocyte cell line by genome-wide gene expression. Osteoarthr. Cartil. 2005, 13, 426–438. [Google Scholar] [CrossRef]
- Redini, F.; Mauviel, A.; Pronost, S.; Loyau, G.; Pujol, J.P. Transforming growth factor beta exerts opposite effects from interleukin-1 beta on cultured rabbit articular chondrocytes through reduction of interleukin-1 receptor expression. Arthritis Rheum 1993, 36, 44–50. [Google Scholar] [CrossRef]
- Glansbeek, H.L.; van Beuningen, H.M.; Vitters, E.L.; van der Kraan, P.M.; van den Berg, W.B. Stimulation of articular cartilage repair in established arthritis by local administration of transforming growth factor-beta into murine knee joints. Lab. Investig. 1998, 78, 133–142. [Google Scholar]
- Pronost, S.; Segond, N.; Macro, M.; Redini, F.; Penfornis, H.; Jullienne, A.; Moukhtar, M.S.; Pujol, J.P. Modulation of interleukin-1 receptor expression by transforming growth factor-beta in cultured rabbit articular chondrocytes: analysis by reverse transcription-polymerase chain reaction. Osteoarthr. Cartil. 1995, 3, 147–155. [Google Scholar] [CrossRef]
- Wiegertjes, R.; van Caam, A.; van Beuningen, H.; Koenders, M.; van Lent, P.; van der Kraan, P.; van de Loo, F.; Blaney Davidson, E. TGF-beta dampens IL-6 signaling in articular chondrocytes by decreasing IL-6 receptor expression. Osteoarthr. Cartil. 2019. [Google Scholar]
- Shiou, S.R.; Yu, Y.; Guo, Y.; Westerhoff, M.; Lu, L.; Petrof, E.O.; Sun, J.; Claud, E.C. Oral administration of transforming growth factor-beta1 (TGF-beta1) protects the immature gut from injury via Smad protein-dependent suppression of epithelial nuclear factor kappaB (NF-kappaB) signaling and proinflammatory cytokine production. J. Biol. Chem. 2013, 288, 34757–34766. [Google Scholar] [CrossRef]
- DiChiara, M.R.; Kiely, J.M.; Gimbrone, M.A., Jr.; Lee, M.E.; Perrella, M.A.; Topper, J.N. Inhibition of E-selectin gene expression by transforming growth factor beta in endothelial cells involves coactivator integration of Smad and nuclear factor kappaB-mediated signals. J. Exp. Med. 2000, 192, 695–704. [Google Scholar] [CrossRef]
- Li, T.F.; Darowish, M.; Zuscik, M.J.; Chen, D.; Schwarz, E.M.; Rosier, R.N.; Drissi, H.; O’Keefe, R.J. Smad3-deficient chondrocytes have enhanced BMP signaling and accelerated differentiation. J. Bone Miner. Res. 2006, 21, 4–16. [Google Scholar] [CrossRef]
- Shen, J.; Li, J.; Wang, B.; Jin, H.; Wang, M.; Zhang, Y.; Yang, Y.; Im, H.J.; O’Keefe, R.; Chen, D. Deletion of the transforming growth factor beta receptor type II gene in articular chondrocytes leads to a progressive osteoarthritis-like phenotype in mice. Arthritis Rheum 2013, 65, 3107–3119. [Google Scholar] [CrossRef]
- Serra, R.; Johnson, M.; Filvaroff, E.H.; LaBorde, J.; Sheehan, D.M.; Derynck, R.; Moses, H.L. Expression of a truncated, kinase-defective TGF-beta type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J. Cell Biol. 1997, 139, 541–552. [Google Scholar] [CrossRef]
- Sueyoshi, T.; Yamamoto, K.; Akiyama, H. Conditional deletion of Tgfbr2 in hypertrophic chondrocytes delays terminal chondrocyte differentiation. Matrix Biol. 2012, 31, 352–359. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Mian, M.; Fu, M.; Zhao, J.Y.; Yang, L.; Li, Y.; Xu, L. Attenuation of the progression of articular cartilage degeneration by inhibition of TGF-beta1 signaling in a mouse model of osteoarthritis. Am. J. Pathol. 2015, 185, 2875–2885. [Google Scholar] [CrossRef]
- Mitsugi, S.; Ariyoshi, W.; Okinaga, T.; Kaneuji, T.; Kataoka, Y.; Takahashi, T.; Nishihara, T. Mechanisms involved in inhibition of chondrogenesis by activin-A. Biochem. Biophys. Res. Commun. 2012, 420, 380–384. [Google Scholar] [CrossRef]
- Hermansson, M.; Sawaji, Y.; Bolton, M.; Alexander, S.; Wallace, A.; Begum, S.; Wait, R.; Saklatvala, J. Proteomic analysis of articular cartilage shows increased type II collagen synthesis in osteoarthritis and expression of inhibin betaA (activin A), a regulatory molecule for chondrocytes. J. Biol. Chem. 2004, 279, 43514–43521. [Google Scholar] [CrossRef]
- Alexander, S.; Watt, F.; Sawaji, Y.; Hermansson, M.; Saklatvala, J. Activin A is an anticatabolic autocrine cytokine in articular cartilage whose production is controlled by fibroblast growth factor 2 and NF-kappaB. Arthritis Rheum 2007, 56, 3715–3725. [Google Scholar] [CrossRef]
- Tardif, G.; Hum, D.; Pelletier, J.P.; Boileau, C.; Ranger, P.; Martel-Pelletier, J. Differential gene expression and regulation of the bone morphogenetic protein antagonists follistatin and gremlin in normal and osteoarthritic human chondrocytes and synovial fibroblasts. Arthritis Rheum 2004, 50, 2521–2530. [Google Scholar] [CrossRef]
- Tardif, G.; Pelletier, J.P.; Boileau, C.; Martel-Pelletier, J. The BMP antagonists follistatin and gremlin in normal and early osteoarthritic cartilage: An immunohistochemical study. Osteoarthr. Cartil. 2009, 17, 263–270. [Google Scholar] [CrossRef]
- Flechtenmacher, J.; Huch, K.; Thonar, E.J.; Mollenhauer, J.A.; Davies, S.R.; Schmid, T.M.; Puhl, W.; Sampath, T.K.; Aydelotte, M.B.; Kuettner, K.E. Recombinant human osteogenic protein 1 is a potent stimulator of the synthesis of cartilage proteoglycans and collagens by human articular chondrocytes. Arthritis Rheum 1996, 39, 1896–1904. [Google Scholar] [CrossRef]
- Luyten, F.P.; Chen, P.; Paralkar, V.; Reddi, A.H. Recombinant bone morphogenetic protein-4, transforming growth factor-beta 1, and activin A enhance the cartilage phenotype of articular chondrocytes in vitro. Exp. Cell Res. 1994, 210, 224–229. [Google Scholar] [CrossRef]
- Chen, A.L.; Fang, C.; Liu, C.; Leslie, M.P.; Chang, E.; Di Cesare, P.E. Expression of bone morphogenetic proteins, receptors, and tissue inhibitors in human fetal, adult, and osteoarthritic articular cartilage. J. Orthop. Res. 2004, 22, 1188–1192. [Google Scholar] [CrossRef]
- van Caam, A.; Madej, W.; Thijssen, E.; Garcia de Vinuesa, A.; van den Berg, W.; Goumans, M.J.; Ten Dijke, P.; Blaney Davidson, E.; van der Kraan, P.M. Expression of TGFbeta-family signalling components in ageing cartilage: Age-related loss of TGFbeta and BMP receptors. Osteoarthr. Cartil. 2016. [Google Scholar]
- Bragdon, B.; Moseychuk, O.; Saldanha, S.; King, D.; Julian, J.; Nohe, A. Bone morphogenetic proteins: A critical review. Cell Signal 2011, 23, 609–620. [Google Scholar] [CrossRef]
- Sengle, G.; Ono, R.N.; Lyons, K.M.; Bachinger, H.P.; Sakai, L.Y. A new model for growth factor activation: Type II receptors compete with the prodomain for BMP-7. J. Mol. Biol. 2008, 381, 1025–1039. [Google Scholar] [CrossRef]
- Aykul, S.; Martinez-Hackert, E. Transforming Growth Factor-beta Family Ligands Can Function as Antagonists by Competing for Type II Receptor Binding. J. Biol. Chem. 2016, 291, 10792–10804. [Google Scholar] [CrossRef]
- Pregizer, S.K.; Mortlock, D.P. Dynamics and cellular localization of Bmp2, Bmp4, and Noggin transcription in the postnatal mouse skeleton. J. Bone Miner. Res. 2015, 30, 64–70. [Google Scholar] [CrossRef]
- Hills, R.L.; Belanger, L.M.; Morris, E.A. Bone morphogenetic protein 9 is a potent anabolic factor for juvenile bovine cartilage, but not adult cartilage. J. Orthop. Res. 2005, 23, 611–617. [Google Scholar] [CrossRef]
- Blaney Davidson, E.N.; Vitters, E.L.; van Lent, P.L.; van de Loo, F.A.; van den Berg, W.B.; van der Kraan, P.M. Elevated extracellular matrix production and degradation upon bone morphogenetic protein-2 (BMP-2) stimulation point toward a role for BMP-2 in cartilage repair and remodeling. Arthritis Res. Ther. 2007, 9, R102. [Google Scholar] [CrossRef]
- Zhou, N.; Li, Q.; Lin, X.; Hu, N.; Liao, J.Y.; Lin, L.B.; Zhao, C.; Hu, Z.M.; Liang, X.; Xu, W.; et al. BMP2 induces chondrogenic differentiation, osteogenic differentiation and endochondral ossification in stem cells. Cell Tissue Res. 2016. [Google Scholar] [CrossRef]
- Blaney Davidson, E.N.; Vitters, E.L.; Bennink, M.B.; van Lent, P.L.; van Caam, A.P.; Blom, A.B.; van den Berg, W.B.; van de Loo, F.A.; van der Kraan, P.M. Inducible chondrocyte-specific overexpression of BMP2 in young mice results in severe aggravation of osteophyte formation in experimental OA without altering cartilage damage. Ann. Rheum. Dis. 2015, 74, 1257–1264. [Google Scholar] [CrossRef]
- Kaneko, K.; Higuchi, C.; Kunugiza, Y.; Yoshida, K.; Sakai, T.; Yoshikawa, H.; Nakata, K. Hyaluronan inhibits BMP-induced osteoblast differentiation. FEBS Lett. 2015, 589, 447–454. [Google Scholar] [CrossRef] [Green Version]
- Klammert, U.; Mueller, T.D.; Hellmann, T.V.; Wuerzler, K.K.; Kotzsch, A.; Schliermann, A.; Schmitz, W.; Kuebler, A.C.; Sebald, W.; Nickel, J. GDF-5 can act as a context-dependent BMP-2 antagonist. BMC Biol. 2015, 13, 77. [Google Scholar] [CrossRef]
- Chang, S.C.; Hoang, B.; Thomas, J.T.; Vukicevic, S.; Luyten, F.P.; Ryba, N.J.; Kozak, C.A.; Reddi, A.H.; Moos, M., Jr. Cartilage-derived morphogenetic proteins. New members of the transforming growth factor-beta superfamily predominantly expressed in long bones during human embryonic development. J. Biol. Chem. 1994, 269, 28227–28234. [Google Scholar]
- Erlacher, L.; Ng, C.K.; Ullrich, R.; Krieger, S.; Luyten, F.P. Presence of cartilage-derived morphogenetic proteins in articular cartilage and enhancement of matrix replacement in vitro. Arthritis Rheum 1998, 41, 263–273. [Google Scholar] [CrossRef]
- Bobacz, K.; Gruber, R.; Soleiman, A.; Graninger, W.B.; Luyten, F.P.; Erlacher, L. Cartilage-derived morphogenetic protein-1 and -2 are endogenously expressed in healthy and osteoarthritic human articular chondrocytes and stimulate matrix synthesis. Osteoarthr. Cartil. 2002, 10, 394–401. [Google Scholar] [CrossRef] [Green Version]
- Chubinskaya, S.; Merrihew, C.; Cs-Szabo, G.; Mollenhauer, J.; McCartney, J.; Rueger, D.C.; Kuettner, K.E. Human articular chondrocytes express osteogenic protein-1. J. Histochem. Cytochem. 2000, 48, 239–250. [Google Scholar] [CrossRef]
- Muehleman, C.; Kuettner, K.E.; Rueger, D.C.; Ten Dijke, P.; Chubinskaya, S. Immunohistochemical localization of osteogenetic protein (OP-1) and its receptors in rabbit articular cartilage. J. Histochem. Cytochem. 2002, 50, 1341–1350. [Google Scholar] [CrossRef]
- Peterson, R.S.; Andhare, R.A.; Rousche, K.T.; Knudson, W.; Wang, W.; Grossfield, J.B.; Thomas, R.O.; Hollingsworth, R.E.; Knudson, C.B. CD44 modulates Smad1 activation in the BMP-7 signaling pathway. J. Cell Biol. 2004, 166, 1081–1091. [Google Scholar] [CrossRef]
- Andhare, R.A.; Takahashi, N.; Knudson, W.; Knudson, C.B. Hyaluronan promotes the chondrocyte response to BMP-7. Osteoarthr. Cartil. 2009, 17, 906–916. [Google Scholar] [CrossRef] [Green Version]
- Luo, N.; Knudson, W.; Askew, E.B.; Veluci, R.; Knudson, C.B. CD44 and hyaluronan promote the bone morphogenetic protein 7 signaling response in murine chondrocytes. Arthritis Rheumatol. 2014, 66, 1547–1558. [Google Scholar] [CrossRef]
- Soder, S.; Hakimiyan, A.; Rueger, D.C.; Kuettner, K.E.; Aigner, T.; Chubinskaya, S. Antisense inhibition of osteogenic protein 1 disturbs human articular cartilage integrity. Arthritis Rheum 2005, 52, 468–478. [Google Scholar] [CrossRef]
- Nishida, Y.; Knudson, C.B.; Eger, W.; Kuettner, K.E.; Knudson, W. Osteogenic protein 1 stimulates cells-associated matrix assembly by normal human articular chondrocytes: up-regulation of hyaluronan synthase, CD44, and aggrecan. Arthritis Rheum 2000, 43, 206–214. [Google Scholar] [CrossRef]
- Huch, K.; Wilbrink, B.; Flechtenmacher, J.; Koepp, H.E.; Aydelotte, M.B.; Sampath, T.K.; Kuettner, K.E.; Mollenhauer, J.; Thonar, E.J. Effects of recombinant human osteogenic protein 1 on the production of proteoglycan, prostaglandin E2, and interleukin-1 receptor antagonist by human articular chondrocytes cultured in the presence of interleukin-1beta. Arthritis Rheum 1997, 40, 2157–2161. [Google Scholar] [CrossRef]
- Chubinskaya, S.; Segalite, D.; Pikovsky, D.; Hakimiyan, A.A.; Rueger, D.C. Effects induced by BMPS in cultures of human articular chondrocytes: Comparative studies. Growth Factors 2008, 26, 275–283. [Google Scholar] [CrossRef]
- Bobacz, K.; Sunk, I.G.; Hofstaetter, J.G.; Amoyo, L.; Toma, C.D.; Akira, S.; Weichhart, T.; Saemann, M.; Smolen, J.S. Toll-like receptors and chondrocytes: The lipopolysaccharide-induced decrease in cartilage matrix synthesis is dependent on the presence of toll-like receptor 4 and antagonized by bone morphogenetic protein 7. Arthritis Rheum 2007, 56, 1880–1893. [Google Scholar] [CrossRef]
- Im, H.J.; Pacione, C.; Chubinskaya, S.; Van Wijnen, A.J.; Sun, Y.; Loeser, R.F. Inhibitory effects of insulin-like growth factor-1 and osteogenic protein-1 on fibronectin fragment- and interleukin-1beta-stimulated matrix metalloproteinase-13 expression in human chondrocytes. J. Biol. Chem. 2003, 278, 25386–25394. [Google Scholar] [CrossRef]
- Abula, K.; Muneta, T.; Miyatake, K.; Yamada, J.; Matsukura, Y.; Inoue, M.; Sekiya, I.; Graf, D.; Economides, A.N.; Rosen, V.; et al. Elimination of BMP7 from the developing limb mesenchyme leads to articular cartilage degeneration and synovial inflammation with increased age. FEBS Lett. 2015, 589, 1240–1248. [Google Scholar] [CrossRef] [Green Version]
- Hurtig, M.; Chubinskaya, S.; Dickey, J.; Rueger, D. BMP-7 protects against progression of cartilage degeneration after impact injury. J. Orthop. Res. 2009, 27, 602–611. [Google Scholar] [CrossRef]
- Badlani, N.; Inoue, A.; Healey, R.; Coutts, R.; Amiel, D. The protective effect of OP-1 on articular cartilage in the development of osteoarthritis. Osteoarthr. Cartil. 2008, 16, 600–606. [Google Scholar] [CrossRef]
- Hayashi, M.; Muneta, T.; Ju, Y.J.; Mochizuki, T.; Sekiya, I. Weekly intra-articular injections of bone morphogenetic protein-7 inhibits osteoarthritis progression. Arthritis Res. Ther. 2008, 10, R118. [Google Scholar] [CrossRef]
- Kuo, A.C.; Rodrigo, J.J.; Reddi, A.H.; Curtiss, S.; Grotkopp, E.; Chiu, M. Microfracture and bone morphogenetic protein 7 (BMP-7) synergistically stimulate articular cartilage repair. Osteoarthr. Cartil. 2006, 14, 1126–1135. [Google Scholar] [CrossRef] [Green Version]
- Spiro, A.S.; Beil, F.T.; Baranowsky, A.; Barvencik, F.; Schilling, A.F.; Nguyen, K.; Khadem, S.; Seitz, S.; Rueger, J.M.; Schinke, T.; et al. BMP-7-induced ectopic bone formation and fracture healing is impaired by systemic NSAID application in C57BL/6-mice. J. Orthop. Res. 2010, 28, 785–791. [Google Scholar] [CrossRef]
- Hunter, D.J.; Pike, M.C.; Jonas, B.L.; Kissin, E.; Krop, J.; McAlindon, T. Phase 1 safety and tolerability study of BMP-7 in symptomatic knee osteoarthritis. BMC Musculoskelet Disord. 2010, 11, 232. [Google Scholar] [CrossRef]
- Chen, P.; Vukicevic, S.; Sampath, T.K.; Luyten, F.P. Bovine articular chondrocytes do not undergo hypertrophy when cultured in the presence of serum and osteogenic protein-1. Biochem. Biophys. Res. Commun. 1993, 197, 1253–1259. [Google Scholar] [CrossRef]
- Caron, M.M.; Emans, P.J.; Cremers, A.; Surtel, D.A.; Coolsen, M.M.; van Rhijn, L.W.; Welting, T.J. Hypertrophic differentiation during chondrogenic differentiation of progenitor cells is stimulated by BMP-2 but suppressed by BMP-7. Osteoarthr. Cartil. 2013, 21, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Haaijman, A.; D’Souza, R.N.; Bronckers, A.L.; Goei, S.W.; Burger, E.H. OP-1 (BMP-7) affects mRNA expression of type I, II, X collagen, and matrix Gla protein in ossifying long bones in vitro. J. Bone Miner. Res. 1997, 12, 1815–1823. [Google Scholar] [CrossRef]
- Caron, M.M.; Emans, P.J.; Surtel, D.A.; van der Kraan, P.M.; van Rhijn, L.W.; Welting, T.J. BAPX-1/NKX-3.2 acts as a chondrocyte hypertrophy molecular switch in osteoarthritis. Arthritis Rheumatol. 2015, 67, 2944–2956. [Google Scholar] [CrossRef]
- Erlacher, L.; McCartney, J.; Piek, E.; ten Dijke, P.; Yanagishita, M.; Oppermann, H.; Luyten, F.P. Cartilage-derived morphogenetic proteins and osteogenic protein-1 differentially regulate osteogenesis. J. Bone Miner. Res. 1998, 13, 383–392. [Google Scholar] [CrossRef]
- Chen, P.; Vukicevic, S.; Sampath, T.K.; Luyten, F.P. Osteogenic protein-1 promotes growth and maturation of chick sternal chondrocytes in serum-free cultures. J. Cell Sci. 1995, 108 Pt 1, 105–114. [Google Scholar]
- Bobacz, K.; Gruber, R.; Soleiman, A.; Erlacher, L.; Smolen, J.S.; Graninger, W.B. Expression of bone morphogenetic protein 6 in healthy and osteoarthritic human articular chondrocytes and stimulation of matrix synthesis in vitro. Arthritis Rheum 2003, 48, 2501–2508. [Google Scholar] [CrossRef]
- Nilsson, O.; Parker, E.A.; Hegde, A.; Chau, M.; Barnes, K.M.; Baron, J. Gradients in bone morphogenetic protein-related gene expression across the growth plate. J. Endocrinol. 2007, 193, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Ito, H.; Akiyama, H.; Shigeno, C.; Nakamura, T. Bone morphogenetic protein-6 and parathyroid hormone-related protein coordinately regulate the hypertrophic conversion in mouse clonal chondrogenic EC cells, ATDC5. Biochim. Biophys. Acta 1999, 1451, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Bidart, M.; Ricard, N.; Levet, S.; Samson, M.; Mallet, C.; David, L.; Subileau, M.; Tillet, E.; Feige, J.J.; Bailly, S. BMP9 is produced by hepatocytes and circulates mainly in an active mature form complexed to its prodomain. Cell Mol. Life Sci. 2012, 69, 313–324. [Google Scholar]
- Blunk, T.; Sieminski, A.L.; Appel, B.; Croft, C.; Courter, D.L.; Chieh, J.J.; Goepferich, A.; Khurana, J.S.; Gooch, K.J. Bone morphogenetic protein 9: A potent modulator of cartilage development in vitro. Growth Factors 2003, 21, 71–77. [Google Scholar] [CrossRef]
- Luu, H.H.; Song, W.X.; Luo, X.; Manning, D.; Luo, J.; Deng, Z.L.; Sharff, K.A.; Montag, A.G.; Haydon, R.C.; He, T.C. Distinct roles of bone morphogenetic proteins in osteogenic differentiation of mesenchymal stem cells. J. Orthop. Res. 2007, 25, 665–677. [Google Scholar] [CrossRef]
- van Caam, A.; Blaney Davidson, E.; Garcia de Vinuesa, A.; van Geffen, E.; van den Berg, W.; Goumans, M.J.; Ten Dijke, P.; van der Kraan, P. The high affinity ALK1-ligand BMP9 induces a hypertrophy-like state in chondrocytes that is antagonized by TGFbeta1. Osteoarthr. Cartil. 2015, 23, 985–995. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Mabuchi, A.; Shi, D.; Kubo, T.; Takatori, Y.; Saito, S.; Fujioka, M.; Sudo, A.; Uchida, A.; Yamamoto, S.; et al. A functional polymorphism in the 5′ UTR of GDF5 is associated with susceptibility to osteoarthritis. Nat. Genet. 2007, 39, 529–533. [Google Scholar] [CrossRef]
- Ratnayake, M.; Ploger, F.; Santibanez-Koref, M.; Loughlin, J. Human chondrocytes respond discordantly to the protein encoded by the osteoarthritis susceptibility gene GDF5. PLoS ONE 2014, 9, e86590. [Google Scholar] [CrossRef]
- Martin, J.A.; Buckwalter, J.A. Aging, articular cartilage chondrocyte senescence and osteoarthritis. Biogerontology 2002, 3, 257–264. [Google Scholar] [CrossRef]
- Chubinskaya, S.; Kumar, B.; Merrihew, C.; Heretis, K.; Rueger, D.C.; Kuettner, K.E. Age-related changes in cartilage endogenous osteogenic protein-1 (OP-1). Biochim. Biophys. Acta 2002, 1588, 126–134. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, J.; Dudhia, J.; Bird, J.L.; Bayliss, M.T. Age-related effects of TGF-beta on proteoglycan synthesis in equine articular cartilage. Biochem. Biophys. Res. Commun. 2000, 274, 467–471. [Google Scholar] [CrossRef]
- Loeser, R.F.; Im, H.J.; Richardson, B.; Lu, Q.; Chubinskaya, S. Methylation of the OP-1 promoter: Potential role in the age-related decline in OP-1 expression in cartilage. Osteoarthr. Cartil. 2009, 17, 513–517. [Google Scholar] [CrossRef]
- Loeser, R.F.; Gandhi, U.; Long, D.L.; Yin, W.; Chubinskaya, S. Aging and oxidative stress reduce the response of human articular chondrocytes to insulin-like growth factor 1 and osteogenic protein 1. Arthritis Rheumatol. 2014, 66, 2201–2209. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, T.; Luo, Q.; Chen, Y.; Leung, V.Y.; Wen, C.; Shah, M.F.; Pan, H.; Chiu, K.; Cao, X.; et al. Cartilage degeneration and excessive subchondral bone formation in spontaneous osteoarthritis involves altered TGF-beta signaling. J. Orthop. Res. 2016, 34, 763–770. [Google Scholar] [CrossRef]
- Bauge, C.; Girard, N.; Lhuissier, E.; Bazille, C.; Boumediene, K. Regulation and Role of TGFbeta Signaling Pathway in Aging and Osteoarthritis Joints. Aging Dis. 2014, 5, 394–405. [Google Scholar]
- Hui, W.; Young, D.A.; Rowan, A.D.; Xu, X.; Cawston, T.E.; Proctor, C.J. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann. Rheum. Dis. 2016, 75, 449–458. [Google Scholar] [CrossRef]
- Hjelmeland, A.B.; Schilling, S.H.; Guo, X.; Quarles, D.; Wang, X.F. Loss of Smad3-mediated negative regulation of Runx2 activity leads to an alteration in cell fate determination. Mol. Cell Biol. 2005, 25, 9460–9468. [Google Scholar] [CrossRef]
- Zheng, L.; Baek, H.J.; Karsenty, G.; Justice, M.J. Filamin B represses chondrocyte hypertrophy in a Runx2/Smad3-dependent manner. J. Cell Biol. 2007, 178, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Responte, D.J.; Lee, J.K.; Hu, J.C.; Athanasiou, K.A. Biomechanics-driven chondrogenesis: From embryo to adult. FASEB J. 2012, 26, 3614–3624. [Google Scholar] [CrossRef]
- Nam, J.; Perera, P.; Rath, B.; Agarwal, S. Dynamic regulation of bone morphogenetic proteins in engineered osteochondral constructs by biomechanical stimulation. Tissue Eng. Part A 2013, 19, 783–792. [Google Scholar] [CrossRef]
- Madej, W.; van Caam, A.; Blaney Davidson, E.; Buma, P.; van der Kraan, P.M. Unloading results in rapid loss of TGFbeta signaling in articular cartilage: Role of loading-induced TGFbeta signaling in maintenance of articular chondrocyte phenotype? Osteoarthr. Cartil. 2016, 24, 1807–1815. [Google Scholar] [CrossRef]
- Hinterwimmer, S.; Krammer, M.; Krotz, M.; Glaser, C.; Baumgart, R.; Reiser, M.; Eckstein, F. Cartilage atrophy in the knees of patients after seven weeks of partial load bearing. Arthritis Rheum 2004, 50, 2516–2520. [Google Scholar] [CrossRef]
- Nomura, M.; Sakitani, N.; Iwasawa, H.; Kohara, Y.; Takano, S.; Wakimoto, Y.; Kuroki, H.; Moriyama, H. Thinning of articular cartilage after joint unloading or immobilization. An experimental investigation of the pathogenesis in mice. Osteoarthr. Cartil. 2017, 25, 727–736. [Google Scholar] [CrossRef]
- Madej, W.; van Caam, A.; Davidson, E.N.; Hannink, G.; Buma, P.; van der Kraan, P.M. Ageing is associated with reduction of mechanically-induced activation of Smad2/3P signaling in articular cartilage. Osteoarthr. Cartil. 2016, 24, 146–157. [Google Scholar] [CrossRef]
- Wallace, I.J.; Worthington, S.; Felson, D.T.; Jurmain, R.D.; Wren, K.T.; Maijanen, H.; Woods, R.J.; Lieberman, D.E. Knee osteoarthritis has doubled in prevalence since the mid-20th century. Proc. Natl. Acad. Sci. USA 2017, 114, 9332–9336. [Google Scholar] [CrossRef] [Green Version]
- Iijima, H.; Ito, A.; Nagai, M.; Tajino, J.; Yamaguchi, S.; Kiyan, W.; Nakahata, A.; Zhang, J.; Wang, T.; Aoyama, T.; et al. Physiological exercise loading suppresses post-traumatic osteoarthritis progression via an increase in bone morphogenetic proteins expression in an experimental rat knee model. Osteoarthr. Cartil. 2017, 25, 964–975. [Google Scholar] [CrossRef]
- Chang, S.H.; Mori, D.; Kobayashi, H.; Mori, Y.; Nakamoto, H.; Okada, K.; Taniguchi, Y.; Sugita, S.; Yano, F.; Chung, U.I.; et al. Excessive mechanical loading promotes osteoarthritis through the gremlin-1-NF-kappaB pathway. Nat. Commun. 2019, 10, 1442. [Google Scholar] [CrossRef]
- Henao-Murillo, L.; Ito, K.; van Donkelaar, C.C. Collagen Damage Location in Articular Cartilage Differs if Damage is Caused by Excessive Loading Magnitude or Rate. Ann. Biomed. Eng. 2018, 46, 605–615. [Google Scholar] [CrossRef] [Green Version]
- Scanzello, C.R.; Goldring, S.R. The role of synovitis in osteoarthritis pathogenesis. Bone 2012, 51, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Robinson, W.H.; Lepus, C.M.; Wang, Q.; Raghu, H.; Mao, R.; Lindstrom, T.M.; Sokolove, J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 580–592. [Google Scholar] [CrossRef]
- D’Agostino, M.A.; Conaghan, P.; Le Bars, M.; Baron, G.; Grassi, W.; Martin-Mola, E.; Wakefield, R.; Brasseur, J.L.; So, A.; Backhaus, M.; et al. EULAR report on the use of ultrasonography in painful knee osteoarthritis. Part 1: Prevalence of inflammation in osteoarthritis. Ann. Rheum. Dis. 2005, 64, 1703–1709. [Google Scholar] [CrossRef]
- Baker, K.; Grainger, A.; Niu, J.; Clancy, M.; Guermazi, A.; Crema, M.; Hughes, L.; Buckwalter, J.; Wooley, A.; Nevitt, M.; et al. Relation of synovitis to knee pain using contrast-enhanced MRIs. Ann. Rheum. Dis. 2010, 69, 1779–1783. [Google Scholar] [CrossRef] [Green Version]
- Sokolove, J.; Lepus, C.M. Role of inflammation in the pathogenesis of osteoarthritis: Latest findings and interpretations. Ther. Adv. Musculoskelet Dis. 2013, 5, 77–94. [Google Scholar] [CrossRef]
- Guermazi, A.; Roemer, F.W.; Hayashi, D.; Crema, M.D.; Niu, J.; Zhang, Y.; Marra, M.D.; Katur, A.; Lynch, J.A.; El-Khoury, G.Y.; et al. Assessment of synovitis with contrast-enhanced MRI using a whole-joint semiquantitative scoring system in people with, or at high risk of, knee osteoarthritis: the MOST study. Ann. Rheum. Dis. 2011, 70, 805–811. [Google Scholar] [CrossRef]
- Pessler, F.; Dai, L.; Diaz-Torne, C.; Gomez-Vaquero, C.; Paessler, M.E.; Zheng, D.H.; Einhorn, E.; Range, U.; Scanzello, C.; Schumacher, H.R. The synovitis of “non-inflammatory” orthopaedic arthropathies: A quantitative histological and immunohistochemical analysis. Ann. Rheum. Dis. 2008, 67, 1184–1187. [Google Scholar] [CrossRef]
- Goldring, M.B.; Goldring, S.R. Osteoarthritis. J. Cell Physiol. 2007, 213, 626–634. [Google Scholar] [CrossRef]
- Bondeson, J.; Blom, A.B.; Wainwright, S.; Hughes, C.; Caterson, B.; van den Berg, W.B. The role of synovial macrophages and macrophage-produced mediators in driving inflammatory and destructive responses in osteoarthritis. Arthritis Rheum 2010, 62, 647–657. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42. [Google Scholar] [CrossRef]
- Wojdasiewicz, P.; Poniatowski, L.A.; Szukiewicz, D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm. 2014, 2014, 561459. [Google Scholar] [CrossRef]
- Sellam, J.; Berenbaum, F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat. Rev. Rheumatol. 2010, 6, 625–635. [Google Scholar] [CrossRef]
- Laavola, M.; Leppanen, T.; Hamalainen, M.; Vuolteenaho, K.; Moilanen, T.; Nieminen, R.; Moilanen, E. IL-6 in Osteoarthritis: Effects of Pine Stilbenoids. Molecules 2018, 24, 109. [Google Scholar] [CrossRef]
- Legendre, F.; Bogdanowicz, P.; Boumediene, K.; Pujol, J.P. Role of interleukin 6 (IL-6)/IL-6R-induced signal tranducers and activators of transcription and mitogen-activated protein kinase/extracellular. J. Rheumatol. 2005, 32, 1307–1316. [Google Scholar]
- Ryu, J.H.; Yang, S.; Shin, Y.; Rhee, J.; Chun, C.H.; Chun, J.S. Interleukin-6 plays an essential role in hypoxia-inducible factor 2alpha-induced experimental osteoarthritic cartilage destruction in mice. Arthritis Rheum 2011, 63, 2732–2743. [Google Scholar] [CrossRef]
- Cortial, D.; Gouttenoire, J.; Rousseau, C.F.; Ronziere, M.C.; Piccardi, N.; Msika, P.; Herbage, D.; Mallein-Gerin, F.; Freyria, A.M. Activation by IL-1 of bovine articular chondrocytes in culture within a 3D collagen-based scaffold. An in vitro model to address the effect of compounds with therapeutic potential in osteoarthritis. Osteoarthr. Cartil. 2006, 14, 631–640. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, J.C.; Martel-Pelletier, J.; Pelletier, J.P. The role of cytokines in osteoarthritis pathophysiology. Biorheology 2002, 39, 237–246. [Google Scholar]
- Bauge, C.; Legendre, F.; Leclercq, S.; Elissalde, J.M.; Pujol, J.P.; Galera, P.; Boumediene, K. Interleukin-1beta impairment of transforming growth factor beta1 signaling by down-regulation of transforming growth factor beta receptor type II and up-regulation of Smad7 in human articular chondrocytes. Arthritis Rheum 2007, 56, 3020–3032. [Google Scholar] [CrossRef]
- Roman-Blas, J.A.; Stokes, D.G.; Jimenez, S.A. Modulation of TGF-beta signaling by proinflammatory cytokines in articular chondrocytes. Osteoarthr. Cartil. 2007, 15, 1367–1377. [Google Scholar] [CrossRef]
- Madej, W.; Buma, P.; van der Kraan, P. Inflammatory conditions partly impair the mechanically mediated activation of Smad2/3 signaling in articular cartilage. Arthritis Res. Ther. 2016, 18, 146. [Google Scholar] [CrossRef] [Green Version]
- Elshaier, A.M.; Hakimiyan, A.A.; Rappoport, L.; Rueger, D.C.; Chubinskaya, S. Effect of interleukin-1beta on osteogenic protein 1-induced signaling in adult human articular chondrocytes. Arthritis Rheum 2009, 60, 143–154. [Google Scholar] [CrossRef]
- Merrihew, C.; Soeder, S.; Rueger, D.C.; Kuettner, K.E.; Chubinskaya, S. Modulation of endogenous osteogenic protein-1 (OP-1) by interleukin-1 in adult human articular cartilage. J. Bone Jt. Surg. Am. 2003, 85-A Suppl 3, 67–74. [Google Scholar] [CrossRef]
- Bauge, C.; Attia, J.; Leclercq, S.; Pujol, J.P.; Galera, P.; Boumediene, K. Interleukin-1beta up-regulation of Smad7 via NF-kappaB activation in human chondrocytes. Arthritis Rheum 2008, 58, 221–226. [Google Scholar] [CrossRef]
- Matsuzaki, K. Smad phospho-isoforms direct context-dependent TGF-beta signaling. Cytokine Growth Factor Rev. 2013, 24, 385–399. [Google Scholar] [CrossRef]
- Kamato, D.; Burch, M.L.; Piva, T.J.; Rezaei, H.B.; Rostam, M.A.; Xu, S.; Zheng, W.; Little, P.J.; Osman, N. Transforming growth factor-beta signalling: Role and consequences of Smad linker region phosphorylation. Cell Signal 2013, 25, 2017–2024. [Google Scholar] [CrossRef]
- Massague, J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef]
- Shi, Y. Structural insights on Smad function in TGFbeta signaling. Bioessays 2001, 23, 223–232. [Google Scholar] [CrossRef]
- Hill, C.S. Nucleocytoplasmic shuttling of Smad proteins. Cell Res. 2009, 19, 36–46. [Google Scholar] [CrossRef]
- Sapkota, G.; Knockaert, M.; Alarcon, C.; Montalvo, E.; Brivanlou, A.H.; Massague, J. Dephosphorylation of the linker regions of Smad1 and Smad2/3 by small C-terminal domain phosphatases has distinct outcomes for bone morphogenetic protein and transforming growth factor-beta pathways. J. Biol. Chem. 2006, 281, 40412–40419. [Google Scholar] [CrossRef]
- Matsuzaki, K. Smad phosphoisoform signaling specificity: The right place at the right time. Carcinogenesis 2011, 32, 1578–1588. [Google Scholar] [CrossRef]
- Millet, C.; Yamashita, M.; Heller, M.; Yu, L.R.; Veenstra, T.D.; Zhang, Y.E. A negative feedback control of transforming growth factor-beta signaling by glycogen synthase kinase 3-mediated Smad3 linker phosphorylation at Ser-204. J. Biol. Chem. 2009, 284, 19808–19816. [Google Scholar] [CrossRef]
- Bae, E.; Sato, M.; Kim, R.J.; Kwak, M.K.; Naka, K.; Gim, J.; Kadota, M.; Tang, B.; Flanders, K.C.; Kim, T.A.; et al. Definition of smad3 phosphorylation events that affect malignant and metastatic behaviors in breast cancer cells. Cancer Res. 2014, 74, 6139–6149. [Google Scholar] [CrossRef]
- Murata, M.; Yoshida, K.; Yamaguchi, T.; Matsuzaki, K. Linker phosphorylation of Smad3 promotes fibro-carcinogenesis in chronic viral hepatitis of hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 15018–15027. [Google Scholar] [CrossRef]
- Demagny, H.; De Robertis, E.M. Point mutations in the tumor suppressor Smad4/DPC4 enhance its phosphorylation by GSK3 and reversibly inactivate TGF-beta signaling. Mol. Cell Oncol 2016, 3, e1025181. [Google Scholar] [CrossRef]
- Demagny, H.; Araki, T.; De Robertis, E.M. The tumor suppressor Smad4/DPC4 is regulated by phosphorylations that integrate FGF, Wnt, and TGF-beta signaling. Cell Rep. 2014, 9, 688–700. [Google Scholar] [CrossRef]
- Heldin, C.H.; Moustakas, A. Role of Smads in TGFbeta signaling. Cell Tissue Res. 2012, 347, 21–36. [Google Scholar] [CrossRef]
- Sekimoto, G.; Matsuzaki, K.; Yoshida, K.; Mori, S.; Murata, M.; Seki, T.; Matsui, H.; Fujisawa, J.; Okazaki, K. Reversible Smad-dependent signaling between tumor suppression and oncogenesis. Cancer Res. 2007, 67, 5090–5096. [Google Scholar] [CrossRef]
- Yu, J.S.; Ramasamy, T.S.; Murphy, N.; Holt, M.K.; Czapiewski, R.; Wei, S.K.; Cui, W. PI3K/mTORC2 regulates TGF-beta/Activin signalling by modulating Smad2/3 activity via linker phosphorylation. Nat. Commun. 2015, 6, 7212. [Google Scholar] [CrossRef]
- Bruce, D.L.; Sapkota, G.P. Phosphatases in SMAD regulation. FEBS Lett. 2012, 586, 1897–1905. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Feng, X.H. Regulation of TGF-beta signalling by protein phosphatases. Biochem. J. 2010, 430, 191–198. [Google Scholar] [CrossRef]
- Saklatvala, J. Inflammatory signaling in cartilage: MAPK and NF-kappaB pathways in chondrocytes and the use of inhibitors for research into pathogenesis and therapy of osteoarthritis. Curr. Drug Targets 2007, 8, 305–313. [Google Scholar] [CrossRef]
- Djouad, F.; Rackwitz, L.; Song, Y.; Janjanin, S.; Tuan, R.S. ERK1/2 activation induced by inflammatory cytokines compromises effective host tissue integration of engineered cartilage. Tissue Eng. Part A 2009, 15, 2825–2835. [Google Scholar] [CrossRef]
- Geng, Y.; Valbracht, J.; Lotz, M. Selective activation of the mitogen-activated protein kinase subgroups c-Jun NH2 terminal kinase and p38 by IL-1 and TNF in human articular chondrocytes. J. Clin. Investig. 1996, 98, 2425–2430. [Google Scholar] [CrossRef]
- Manley, G.C.A.; Stokes, C.A.; Marsh, E.K.; Sabroe, I.; Parker, L.C. DUSP10 Negatively Regulates the Inflammatory Response to Rhinovirus through Interleukin-1beta Signaling. J. Virol. 2019, 93. [Google Scholar]
- Arnold, M.A.; Kim, Y.; Czubryt, M.P.; Phan, J.; McAnally, D.; Qi, X.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev. Cell 2007, 12, 377–389. [Google Scholar] [CrossRef]
- Quinn, Z.A.; Yang, C.C.; Wrana, J.L.; McDermott, J.C. Smad proteins function as co-modulators for MEF2 transcriptional regulatory proteins. Nucleic Acids Res. 2001, 29, 732–742. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Kang, J.S.; Derynck, R. TGF-beta-activated Smad3 represses MEF2-dependent transcription in myogenic differentiation. EMBO J. 2004, 23, 1557–1566. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thielen, N.G.M.; van der Kraan, P.M.; van Caam, A.P.M. TGFβ/BMP Signaling Pathway in Cartilage Homeostasis. Cells 2019, 8, 969. https://doi.org/10.3390/cells8090969
Thielen NGM, van der Kraan PM, van Caam APM. TGFβ/BMP Signaling Pathway in Cartilage Homeostasis. Cells. 2019; 8(9):969. https://doi.org/10.3390/cells8090969
Chicago/Turabian StyleThielen, Nathalie G.M., Peter M. van der Kraan, and Arjan P.M. van Caam. 2019. "TGFβ/BMP Signaling Pathway in Cartilage Homeostasis" Cells 8, no. 9: 969. https://doi.org/10.3390/cells8090969
APA StyleThielen, N. G. M., van der Kraan, P. M., & van Caam, A. P. M. (2019). TGFβ/BMP Signaling Pathway in Cartilage Homeostasis. Cells, 8(9), 969. https://doi.org/10.3390/cells8090969