Somatic Mutations of lats2 Cause Peripheral Nerve Sheath Tumors in Zebrafish

Abstract
:1. Introduction
2. Materials and Methods
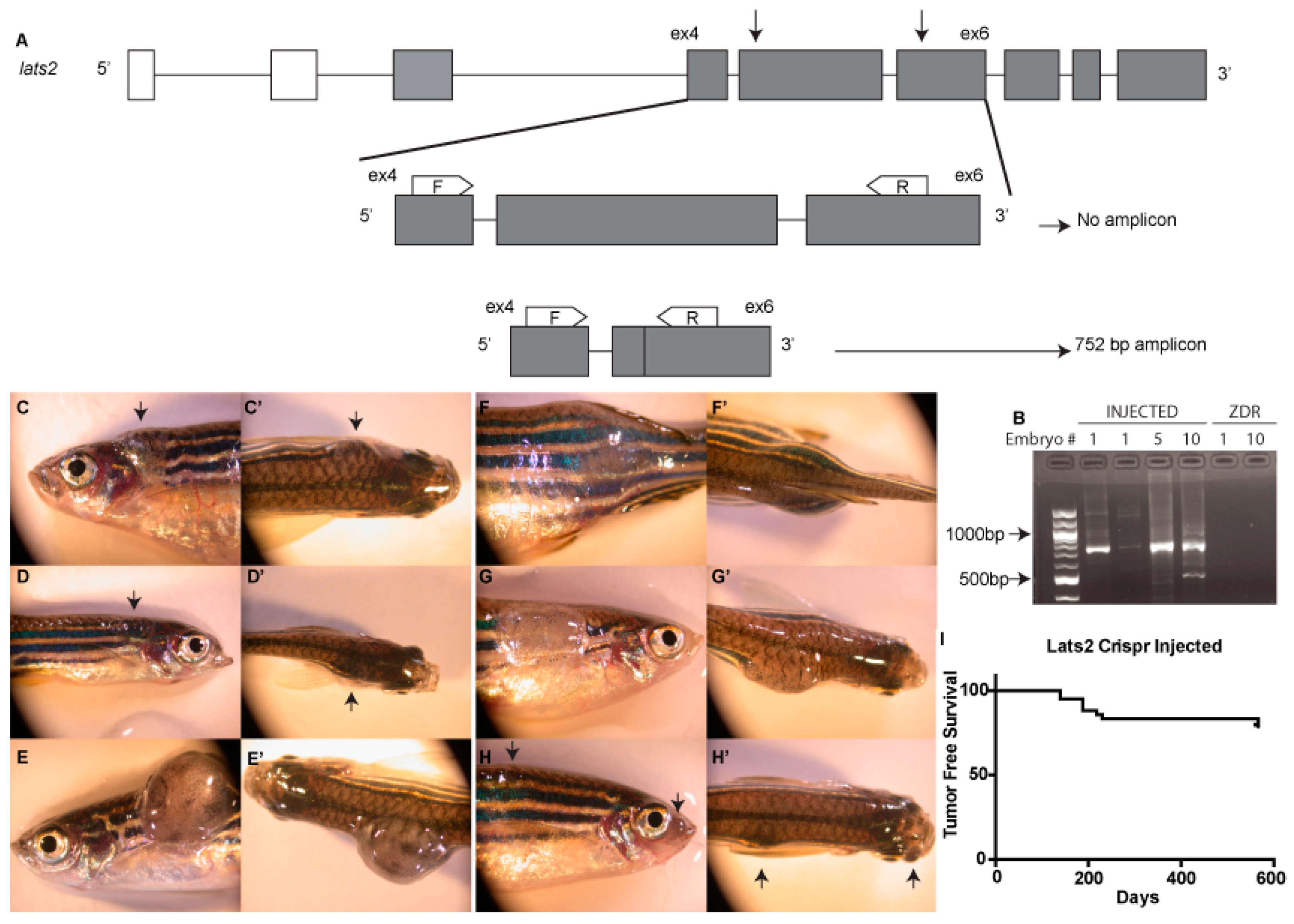
2.1. CRISPR Design
2.2. Genotyping
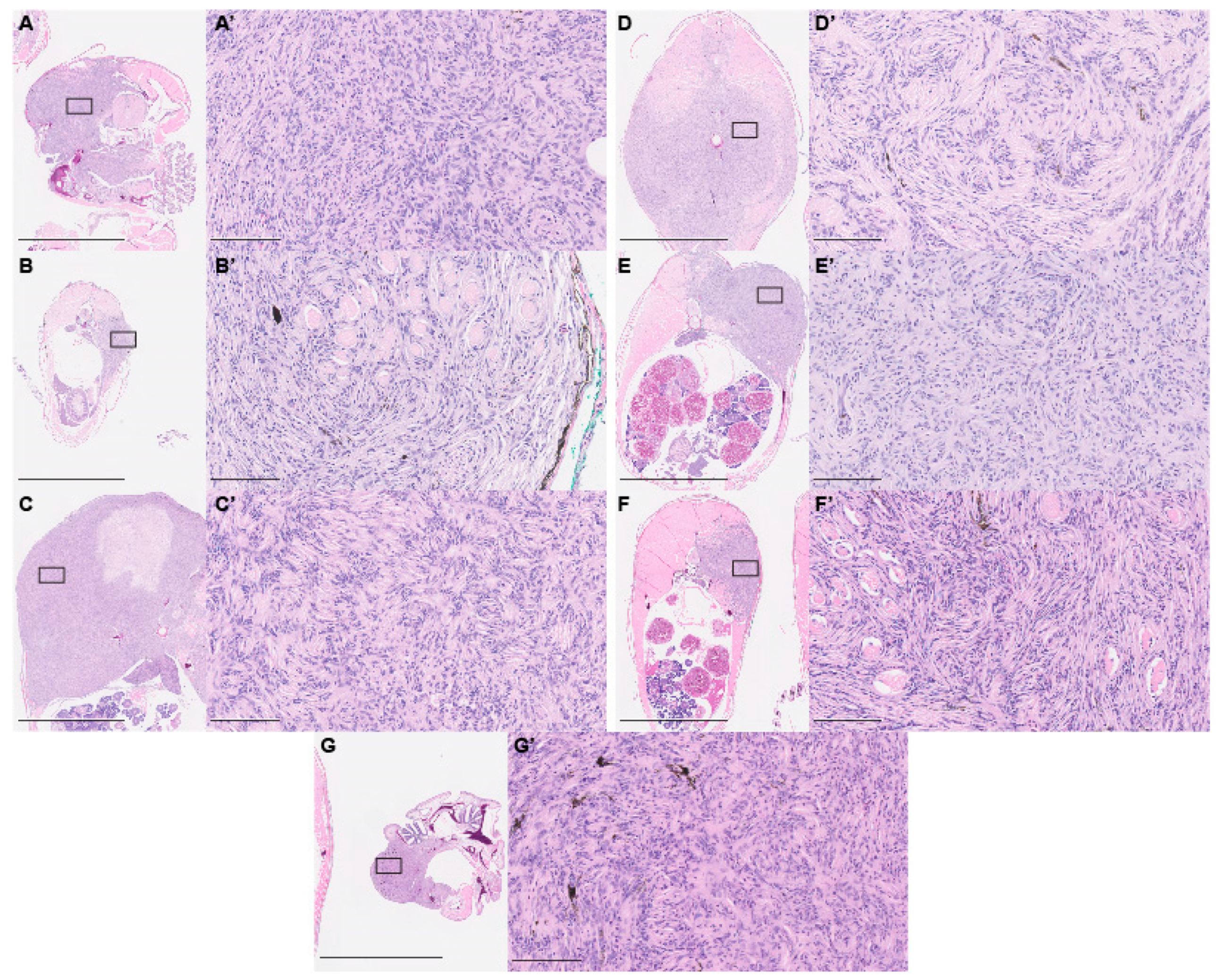
2.3. Paraffin Histology
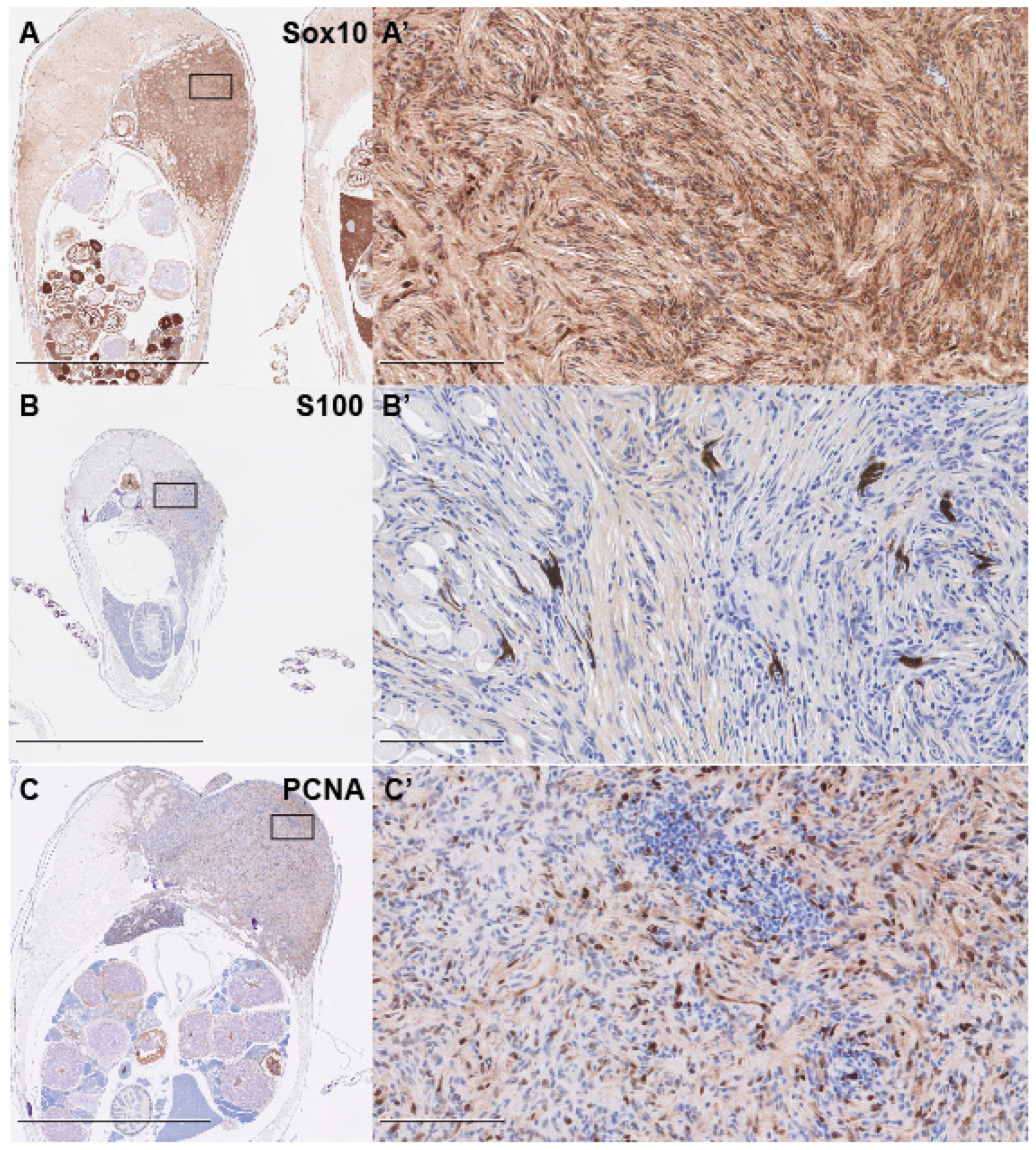
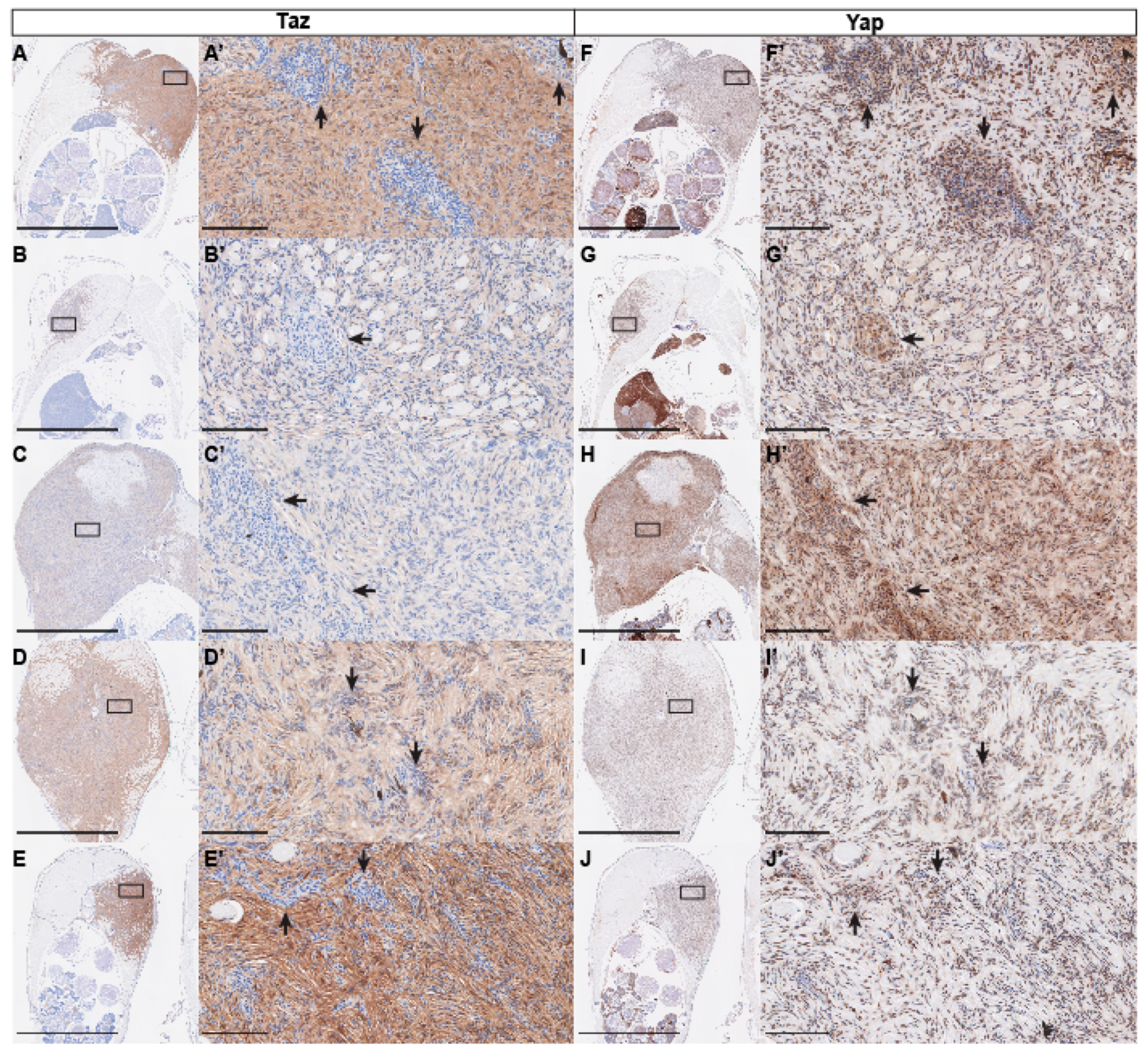
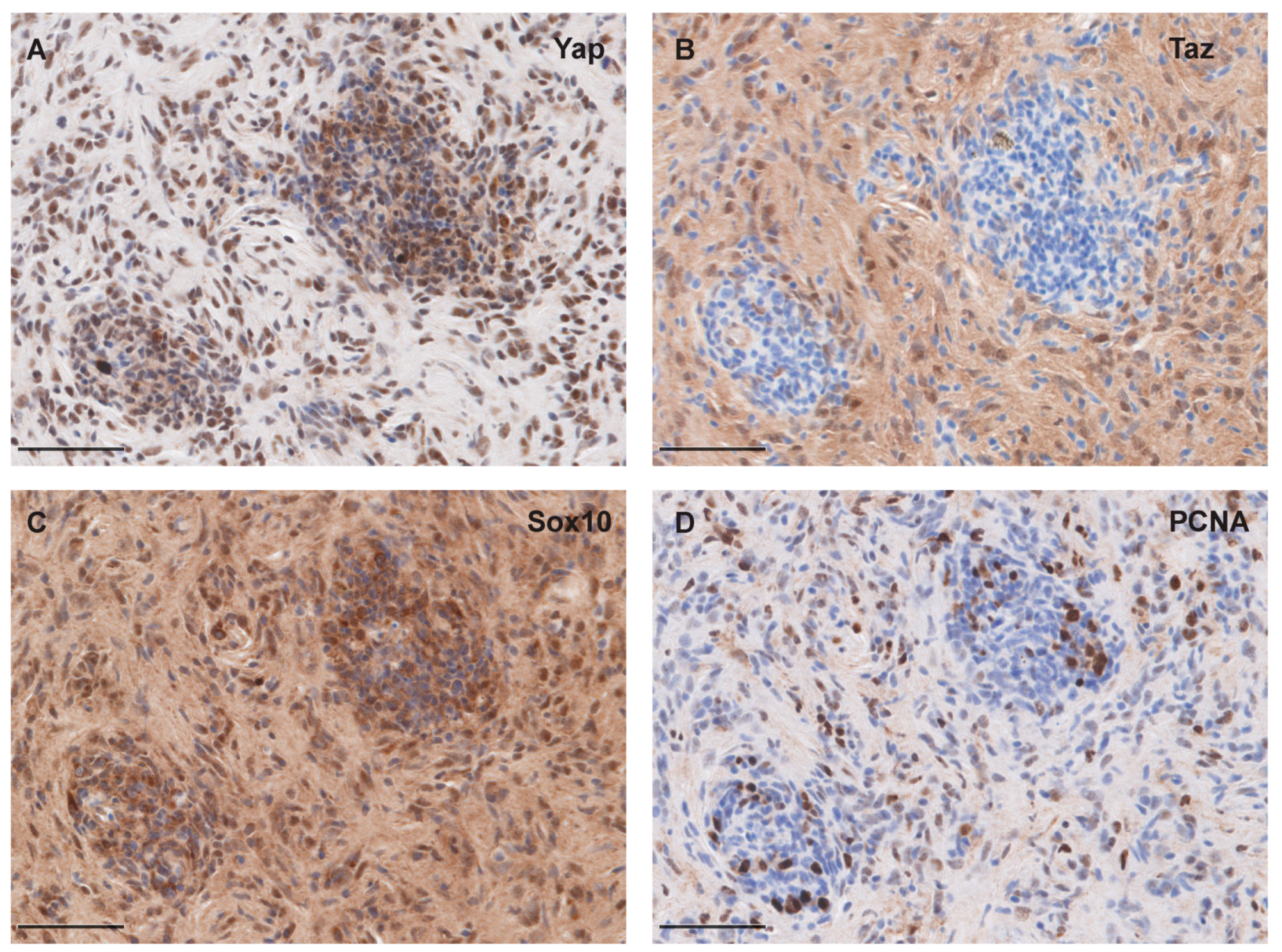
2.4. Immunohistochemistry
2.5. Nomenclature
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro-Oncology 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, T. The 2016 WHO Classification of Tumours of the Central Nervous System: The Major Points of Revision. Neurol. Medico-Chirurgica 2017, 57, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sughrue, M.E.; Levine, J.; Barbaro, N.M. Pain as a symptom of peripheral nerve sheath tumors: Clinical significance and future therapeutic directions. J. Brachial Plex. Peripher. Nerve Inj. 2008, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Bradford, D.; Kim, A. Current Treatment Options for Malignant Peripheral Nerve Sheath Tumors. Curr. Treat. Options Oncol. 2015, 16, 12. [Google Scholar] [CrossRef] [PubMed]
- James, A.W.; Shurell, E.; Singh, A.; Dry, S.M.; Eilber, F.C. Malignant Peripheral Nerve Sheath Tumor. Surg. Oncol. Clin. 2016, 25, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Carroll, S.L. Molecular mechanisms promoting the pathogenesis of Schwann cell neoplasms. Acta Neuropathol. 2012, 123, 321–348. [Google Scholar] [CrossRef] [PubMed]
- Korf, B.R. Pediatric Neurology Part I: Chapter 39—Neurofibromatosis. In Handbook of Clinical Neurology; Dulac, O., Lassonde, M., Sarnat, H.B., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; Volume 111, pp. 333–340. [Google Scholar]
- Messersmith, L.; Krauland, K. Neurofibroma; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Kiuru, M.; Busam, K.J. The NF1 gene in tumor syndromes and melanoma. Lab. Investig. 2017, 97, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Rouleau, G.A.; Merel, P.; Lutchman, M.; Sanson, M.; Zucman, J.; Marineau, C.; Hoang-Xuan, K.; Demczuk, S.; Desmaze, C.; Plougastel, B.; et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature 1993, 363, 515–521. [Google Scholar] [CrossRef]
- Trofatter, J.A.; MacCollin, M.M.; Rutter, J.L.; Murrell, J.R.; Duyao, M.P.; Parry, D.M.; Eldridge, R.; Kley, N.; Menon, A.G.; Pulaski, K. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell 1993, 72, 791–800. [Google Scholar] [CrossRef]
- Hamaratoglu, F.; Willecke, M.; Kango-Singh, M.; Nolo, R.; Hyun, E.; Tao, C.; Jafar-Nejad, H.; Halder, G. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat. Cell Biol. 2006, 8, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Mindos, T.; Dun, X.; North, K.; Doddrell, R.D.S.; Schulz, A.; Edwards, P.; Russell, J.; Gray, B.; Roberts, S.L.; Shivane, A.; et al. Merlin controls the repair capacity of Schwann cells after injury by regulating Hippo/YAP activity. J. Cell Biol. 2017, 216, 495–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farid, M.; Demicco, E.G.; Garcia, R.; Ahn, L.; Merola, P.R.; Cioffi, A.; Maki, R.G. Malignant Peripheral Nerve Sheath Tumors. The Oncologist 2014, 19, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Wu, L.M.N.; Bai, S.; Zhao, C.; Wang, H.; Wang, J.; Xu, L.; Sakabe, M.; Zhou, W.; Xin, M.; et al. A reciprocal regulatory loop between TAZ/YAP and G-protein Gαs regulates Schwann cell proliferation and myelination. Nat. Commun. 2017, 8, 15161. [Google Scholar] [CrossRef] [PubMed]
- Poitelon, Y.; Lopez-Anido, C.; Catignas, K.; Berti, C.; Palmisano, M.; Williamson, C.; Ameroso, D.; Abiko, K.; Hwang, Y.; Gregorieff, A.; et al. YAP and TAZ control peripheral myelination and the expression of laminin receptors in Schwann cells. Nat. Neurosci. 2016, 19, 879–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grove, M.; Kim, H.; Santerre, M.; Krupka, A.J.; Han, S.B.; Zhai, J.; Cho, J.Y.; Park, R.; Harris, M.; Kim, S.; et al. YAP/TAZ initiate and maintain Schwann cell myelination. eLife 2017, 6, e20982. [Google Scholar] [CrossRef]
- Li, W.; Cooper, J.; Zhou, L.; Yang, C.; Erdjument-Bromage, H.; Zagzag, D.; Snuderl, M.; Ladanyi, M.; Hanemann, C.O.; Zhou, P.; et al. Merlin/NF2 Loss-Driven Tumorigenesis Linked to CRL4DCAF1-Mediated Inhibition of the Hippo Pathway Kinases Lats1 and 2 in the Nucleus. Cancer Cell 2014, 26, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Guerrant, W.; Kota, S.; Troutman, S.; Mandati, V.; Fallahi, M.; Stemmer-Rachamimov, A.; Kissil, J.L. YAP Mediates Tumorigenesis in Neurofibromatosis Type 2 by Promoting Cell Survival and Proliferation through a COX-2–EGFR Signaling Axis. Cancer Res. 2016, 76, 3507–3519. [Google Scholar] [CrossRef]
- Boin, A.; Couvelard, A.; Couderc, C.; Brito, I.; Filipescu, D.; Kalamarides, M.; Bedossa, P.; De Koning, L.; Danelsky, C.; Dubois, T.; et al. Proteomic screening identifies a YAP-driven signaling network linked to tumor cell proliferation in human schwannomas. Neuro-Oncology 2014, 16, 1196–1209. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-H.; Ohta, T.; Oh, J.E.; Le Calvez-Kelm, F.; McKay, J.; Voegele, C.; Durand, G.; Mittelbronn, M.; Kleihues, P.; Paulus, W.; et al. TP53, MSH4, and LATS1 Germline Mutations in a Family with Clustering of Nervous System Tumors. Am. J. Pathol. 2014, 184, 2374–2381. [Google Scholar] [CrossRef]
- Oh, J.-E.; Ohta, T.; Satomi, K.; Foll, M.; Durand, G.; McKay, J.; Calvez-Kelm, F.L.; Mittelbronn, M.; Brokinkel, B.; Paulus, W.; et al. Alterations in the NF2/LATS1/LATS2/YAP Pathway in Schwannomas. J. Neuropathol. Exp. Neurol. 2015, 74, 952–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.; Yang, Z.; Chen, Y.; Zhou, Q.; Zhang, J.; Liu, J.; Wang, B.; He, Q.; Zhang, L.; Yu, Y.; et al. Deregulation of the Hippo Pathway Promotes Tumor Cell Proliferation Through YAP Activity in Human Sporadic Vestibular Schwannoma. World Neurosurg. 2018, 117, e269–e279. [Google Scholar] [CrossRef] [PubMed]
- Faden, D.L.; Asthana, S.; Tihan, T.; DeRisi, J.; Kliot, M. Whole Exome Sequencing of Growing and Non-Growing Cutaneous Neurofibromas from a Single Patient with Neurofibromatosis Type 1. PLoS ONE 2017, 12, e0170348. [Google Scholar]
- Tanahashi, K.; Natsume, A.; Ohka, F.; Motomura, K.; Alim, A.; Tanaka, I.; Senga, T.; Harada, I.; Fukuyama, R.; Sumiyoshi, N.; et al. Activation of Yes-Associated Protein in Low-Grade Meningiomas Is Regulated by Merlin, Cell Density, and Extracellular Matrix Stiffness. J. Neuropathol. Exp. Neurol. 2015, 74, 704–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, A.M.; Missiaglia, E.; Galli, G.G.; Hettmer, S.; Urcia, R.; Carrara, M.; Judson, R.N.; Thway, K.; Nadal, G.; Selfe, J.L.; et al. The Hippo Transducer YAP1 Transforms Activated Satellite Cells and Is a Potent Effector of Embryonal Rhabdomyosarcoma Formation. Cancer Cell 2014, 26, 273–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tranchant, R.; Quetel, L.; Tallet, A.; Meiller, C.; Renier, A.; de Koning, L.; de Reynies, A.; Pimpec-Barthes, F.L.; Zucman-Rossi, J.; Jaurand, M.-C.; et al. Co-occurring Mutations of Tumor Suppressor Genes, LATS2 and NF2, in Malignant Pleural Mesothelioma. Clin. Cancer Res. 2017, 23, 3191–3202. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.M.N.; Deng, Y.; Wang, J.; Zhao, C.; Wang, J.; Rao, R.; Xu, L.; Zhou, W.; Choi, K.; Rizvi, T.A.; et al. Programming of Schwann Cells by Lats1/2-TAZ/YAP Signaling Drives Malignant Peripheral Nerve Sheath Tumorigenesis. Cancer Cell 2018, 33, 292–308.e7. [Google Scholar] [CrossRef] [Green Version]
- Bootorabi, F.; Manouchehri, H.; Changizi, R.; Barker, H.; Palazzo, E.; Saltari, A.; Parikka, M.; Pincelli, C.; Aspatwar, A. Zebrafish as a Model Organism for the Development of Drugs for Skin Cancer. Int. J. Mol. Sci. 2017, 18, 1550. [Google Scholar] [CrossRef]
- Ablain, J.; Xu, M.; Rothschild, H.; Jordan, R.C.; Mito, J.K.; Daniels, B.H.; Bell, C.F.; Joseph, N.M.; Wu, H.; Bastian, B.C.; et al. Human tumor genomics and zebrafish modeling identify SPRED1 loss as a driver of mucosal melanoma. Science 2018, 362, 1055–1060. [Google Scholar] [CrossRef]
- Durbin, A.D.; Ki, D.H.; He, S.; Look, A.T. Malignant Peripheral Nerve Sheath Tumors. In Cancer and Zebrafish: Mechanisms, Techniques, and Models; Langenau, D.M., Ed.; Springer International Publishing: New York, NY, USA, 2016; pp. 495–530. ISBN 978-3-319-30654-4. [Google Scholar]
- Ki, D.H.; He, S.; Rodig, S.; Look, A.T. Overexpression of PDGFRA cooperates with loss of NF1 and p53 to accelerate the molecular pathogenesis of malignant peripheral nerve sheath tumors. Oncogene 2017, 36, 1058–1068. [Google Scholar] [CrossRef]
- Solin, S.L.; Shive, H.R.; Woolard, K.D.; Essner, J.J.; McGrail, M. Rapid tumor induction in zebrafish by TALEN-mediated somatic inactivation of the retinoblastoma1 tumor suppressor rb1. Sci. Rep. 2015, 5, 13745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, J.; Choi, J.-H.; Park, M.-H.; Kim, H.; Kim, J.H.; Kim, S.-Y.; Hong, D.; Kim, S.; Lee, J.E.; Kim, C.-H.; et al. Development of zebrafish medulloblastoma-like PNET model by TALEN-mediated somatic gene inactivation. Oncotarget 2017, 8, 55280–55297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, L.E.; Haltom, J.A.; Almeida, M.P.; Wierson, W.A.; Solin, S.L.; Weiss, T.J.; Helmer, J.A.; Sandquist, E.J.; Shive, H.R.; McGrail, M. Epigenetic regulators Rbbp4 and Hdac1 are overexpressed in a zebrafish model of RB1 embryonal brain tumor, and are required for neural progenitor survival and proliferation. Dis. Model. Mech. 2018, 11, dmm034124. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Miyazaki, T.; Chow, R.W.-Y.; Ishikawa, H.; Nakajima, H.; Vermot, J.; Mochizuki, N. Hippo signaling determines the number of venous pole cells that originate from the anterior lateral plate mesoderm in zebrafish. eLife 2018, 7, e29106. [Google Scholar] [CrossRef] [PubMed]
- El-Brolosy, M.A.; Kontarakis, Z.; Rossi, A.; Kuenne, C.; Günther, S.; Fukuda, N.; Kikhi, K.; Boezio, G.L.M.; Takacs, C.M.; Lai, S.-L.; et al. Genetic compensation triggered by mutant mRNA degradation. Nature 2019, 568, 193–197. [Google Scholar] [CrossRef]
- Ma, Z.; Zhu, P.; Shi, H.; Guo, L.; Zhang, Q.; Chen, Y.; Chen, S.; Zhang, Z.; Peng, J.; Chen, J. PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature 2019, 568, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Pekmezci, M.; Reuss, D.E.; Hirbe, A.C.; Dahiya, S.; Gutmann, D.H.; von Deimling, A.; Horvai, A.E.; Perry, A. Morphologic and immunohistochemical features of malignant peripheral nerve sheath tumors and cellular schwannomas. Mod. Pathol. 2015, 28, 187–200. [Google Scholar] [CrossRef]
- Rodriguez, F.J.; Folpe, A.L.; Giannini, C.; Perry, A. Pathology of peripheral nerve sheath tumors: Diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012, 123, 295–319. [Google Scholar] [CrossRef]
- Karamchandani, J.; Nielsen, T.; van de Rijn, M.; West, R. Sox10 and S100 in the Diagnosis of Soft-tissue Neoplasms. Appl. Immunohistochem. Mol. Morphol. 2012, 20, 445–450. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, M.; McCue, P.A.; Sarlomo-Rikala, M.; Biernat, W.; Czapiewski, P.; Kopczynski, J.; Thompson, L.D.; Lasota, J.; Wang, Z.; Fetsch, J.F. Sox10—A marker for not only Schwannian and melanocytic neoplasms but also myoepithelial cell tumors of soft tissue. A systematic analysis of 5134 tumors. Am. J. Surg. Pathol. 2015, 39, 826–835. [Google Scholar] [CrossRef]
- Miesfeld, J.B.; Gestri, G.; Clark, B.S.; Flinn, M.A.; Poole, R.J.; Bader, J.R.; Besharse, J.C.; Wilson, S.W.; Link, B.A. Yap and Taz regulate retinal pigment epithelial cell fate. Development 2015, 142, 3021–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimelman, D.; Smith, N.L.; Lai, J.K.H.; Stainier, D.Y. Regulation of posterior body and epidermal morphogenesis in zebrafish by localized Yap1 and Wwtr1. eLife 2017, 6, e31065. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.K.H.; Collins, M.M.; Uribe, V.; Jiménez-Amilburu, V.; Günther, S.; Maischein, H.-M.; Stainier, D.Y.R. The Hippo pathway effector Wwtr1 regulates cardiac wall maturation in zebrafish. Development 2018, 145, dev159210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, S.-Y.; Kim, M.; Kim, H.-S.; Huh, T.-L.; Chitnis, A.B. Fluorescent protein expression driven by her4 regulatory elements reveals the spatiotemporal pattern of Notch signaling in the nervous system of zebrafish embryos. Dev. Biol. 2007, 301, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, M.J.; Pisharath, H.; Yusuff, S.; Moore, J.C.; Siekmann, A.F.; Lawson, N.; Leach, S.D. Notch-responsive cells initiate the secondary transition in larval zebrafish pancreas. Mech. Dev. 2009, 126, 898–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collery, R.F.; Link, B.A. Dynamic smad-mediated BMP signaling revealed through transgenic zebrafish. Dev. Dyn. 2011, 240, 712–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laux, D.W.; Febbo, J.A.; Roman, B.L. Dynamic analysis of BMP-responsive smad activity in live zebrafish embryos. Dev. Dyn. 2011, 240, 682–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramel, M.-C.; Hill, C.S. The ventral to dorsal BMP activity gradient in the early zebrafish embryo is determined by graded expression of BMP ligands. Dev. Biol. 2013, 378, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Dorsky, R.I.; Sheldahl, L.C.; Moon, R.T. A Transgenic Lef1/β-Catenin-Dependent Reporter Is Expressed in Spatially Restricted Domains throughout Zebrafish Development. Dev. Biol. 2002, 241, 229–237. [Google Scholar] [CrossRef]
- Shimizu, N.; Kawakami, K.; Ishitani, T. Visualization and exploration of Tcf/Lef function using a highly responsive Wnt/β-catenin signaling-reporter transgenic zebrafish. Dev. Biol. 2012, 370, 71–85. [Google Scholar] [CrossRef]
- Mich, J.K.; Payumo, A.Y.; Rack, P.G.; Chen, J.K. In Vivo Imaging of Hedgehog Pathway Activation with a Nuclear Fluorescent Reporter. PLoS ONE 2014, 9, e103661. [Google Scholar] [CrossRef] [PubMed]
- Schwend, T.; Loucks, E.J.; Ahlgren, S.C. Visualization of Gli Activity in Craniofacial Tissues of Hedgehog-Pathway Reporter Transgenic Zebrafish. PLoS ONE 2010, 5, e14396. [Google Scholar] [CrossRef] [PubMed]
- Astone, M.; Lai, J.K.H.; Dupont, S.; Stainier, D.Y.R.; Argenton, F.; Vettori, A. Zebrafish mutants and TEAD reporters reveal essential functions for Yap and Taz in posterior cardinal vein development. Sci. Rep. 2018, 8, 10189. [Google Scholar] [CrossRef] [PubMed]
- Miesfeld, J.B.; Link, B.A. Establishment of transgenic lines to monitor and manipulate Yap/Taz-Tead activity in zebrafish reveals both evolutionarily conserved and divergent functions of the Hippo pathway. Mech. Dev. 2014, 133, 177–188. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| lats2WT/WT | lats2WT/mw87 | lats2mw87/mw87 | |
|---|---|---|---|
| Expected | 14.25 (25%) | 28.50 (50%) | 14.25 (25%) |
| Observed | 19 (33.33%) | 35 (61.40%) | 3 (5.26%) |
| Chi-square | 11.95 | ||
| Degrees Freedom | 2 | ||
| p value (two tailed) | 0.0025 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brandt, Z.J.; North, P.N.; Link, B.A. Somatic Mutations of lats2 Cause Peripheral Nerve Sheath Tumors in Zebrafish. Cells 2019, 8, 972. https://doi.org/10.3390/cells8090972
Brandt ZJ, North PN, Link BA. Somatic Mutations of lats2 Cause Peripheral Nerve Sheath Tumors in Zebrafish. Cells. 2019; 8(9):972. https://doi.org/10.3390/cells8090972
Chicago/Turabian StyleBrandt, Zachary J., Paula N. North, and Brian A. Link. 2019. "Somatic Mutations of lats2 Cause Peripheral Nerve Sheath Tumors in Zebrafish" Cells 8, no. 9: 972. https://doi.org/10.3390/cells8090972
APA StyleBrandt, Z. J., North, P. N., & Link, B. A. (2019). Somatic Mutations of lats2 Cause Peripheral Nerve Sheath Tumors in Zebrafish. Cells, 8(9), 972. https://doi.org/10.3390/cells8090972