Transcriptional Coactivator TAZ Negatively Regulates Tumor Suppressor p53 Activity and Cellular Senescence

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Plasmids
2.3. Antibodies and Reagents
2.4. Luciferase Assay
2.5. Immunoprecipitation and Immunoblotting
2.6. GST Pull-Down Assay
2.7. RNA Isolation and Quantitative PCR (qPCR)
2.8. Chromatin Immunoprecipitation (ChIP) Assay
2.9. Cell Viability Assay, Apoptosis Assay, and Senescence-Associated β-gal (SA-β-gal) Staining
2.10. Statistical Analysis
3. Results
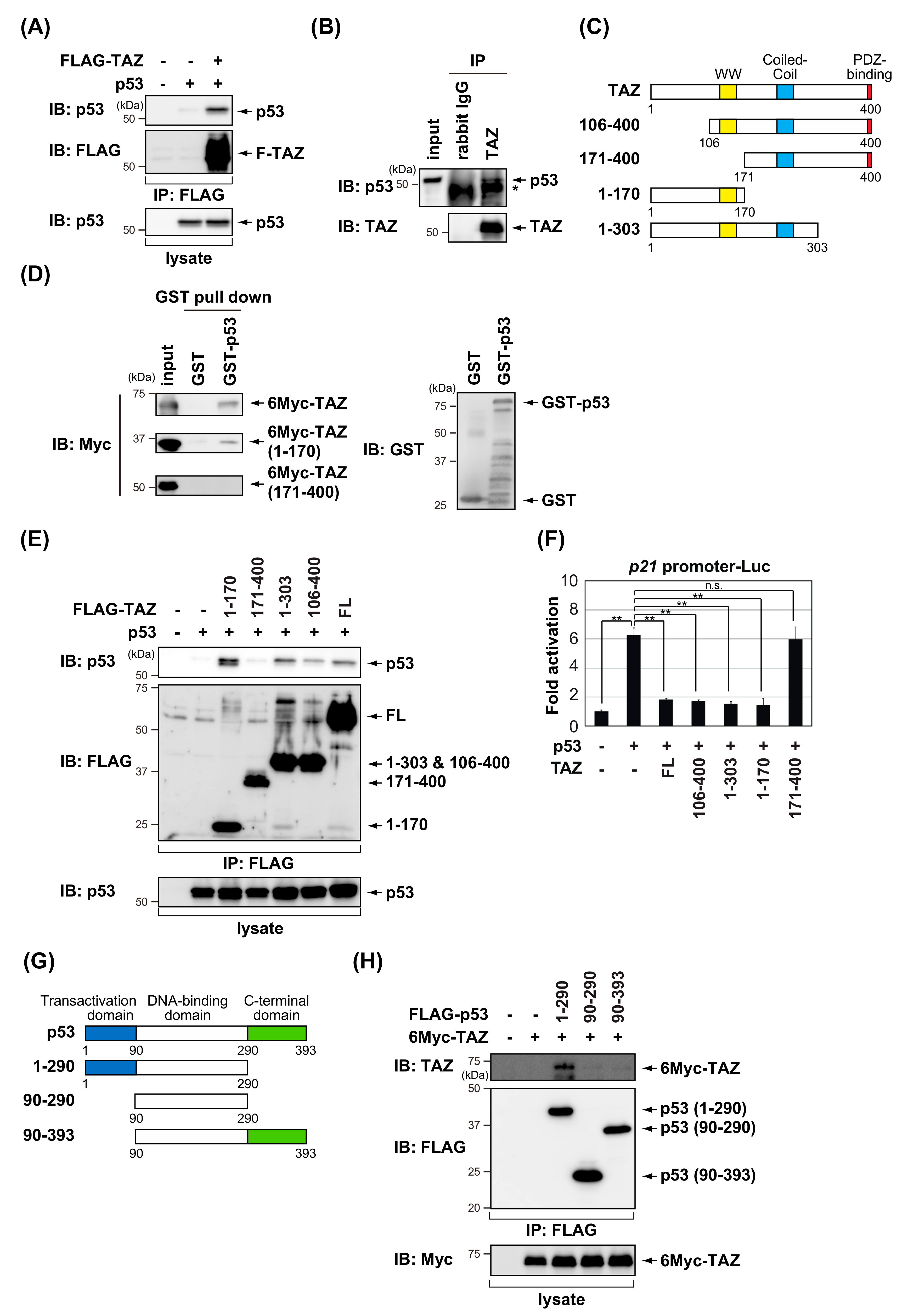
3.1. TAZ Represses the Transcriptional Activity of p53
3.2. TAZ Interacts with p53
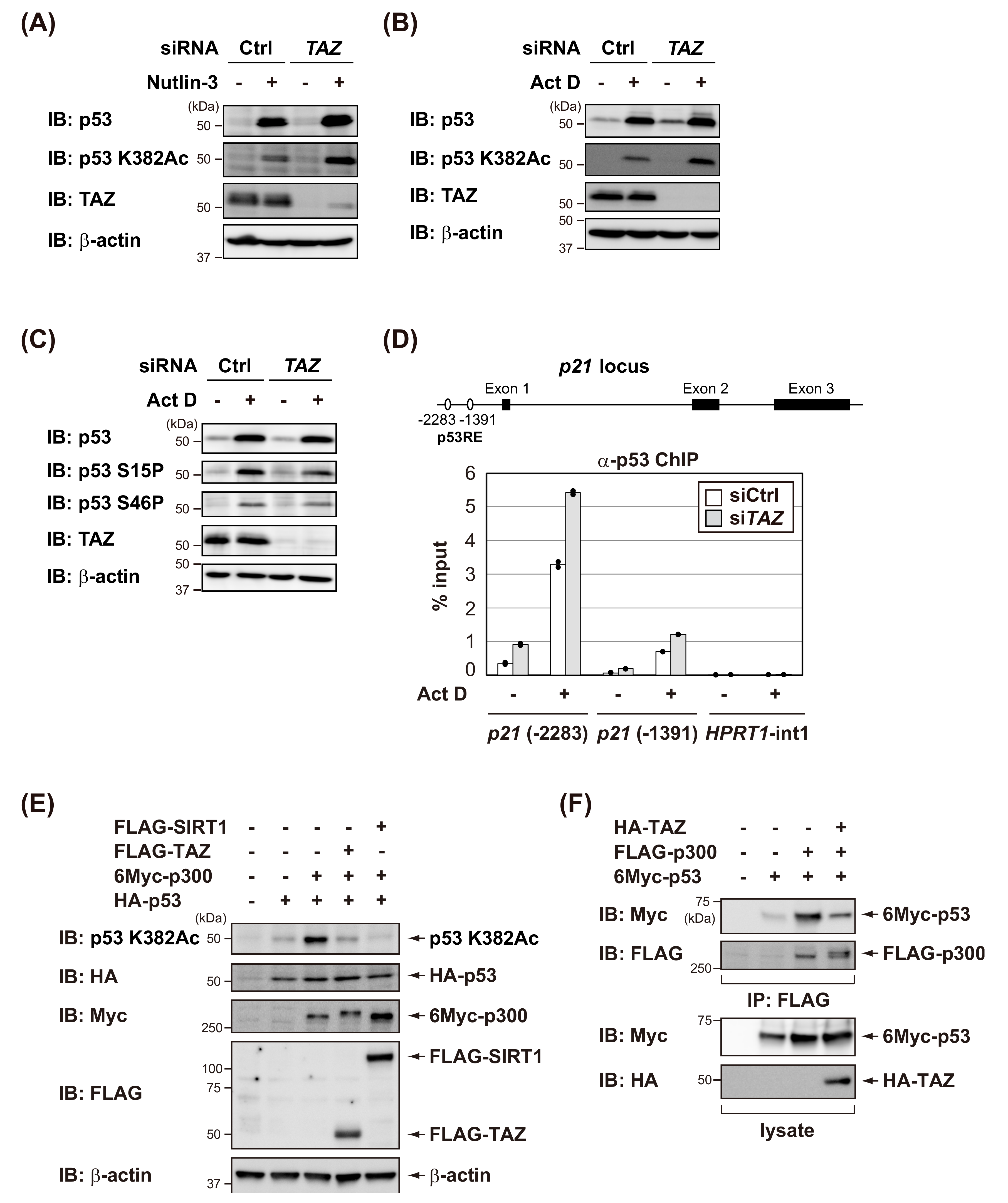
3.3. TAZ Suppresses p300-Mediated Acetylation of p53 and Reduces the DNA-Binding Activity of p53
3.4. TAZ Knockdown Induces p53-Dependent Senescence in Normal Human Fibroblasts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kanai, F.; Mariignani, P.A.; Sarbassova, D.; Yagi, R.; Donowitz, M.; Hisaminato, A.; Fujiwara, T.; Ito, Y.; Cantley, L.C.; Yaffe, M.B. TAZ: A novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 2000, 19, 6778–6791. [Google Scholar] [CrossRef]
- Zhou, X.; Lei, Q.Y. Regulation of TAZ in cancer. Protein Cell 2016, 7, 548–561. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the roots of cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Oren, M. Decision making by p53: Life, death and cancer. Cell Death Differ. 2003, 10, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Prives, C.; Hall, P.A. The p53 pathway. J. Pathol. 1999, 187, 112–126. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Beckerman, R.; Prives, C. Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2010, 2, a000935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is indispensable for p53 activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, C.L.; Gu, W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2011, 2, 456–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, A.; Kawaguchi, Y.; Lai, C.H.; Kovacs, J.J.; Higashimoto, Y.; Appella, E.; Yao, T.P. MDM2-HDAC1-mediated deacetylation of p53 is recruited for its degradation. EMBO J. 2002, 21, 6236–6245. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, C.; Inoue, Y.; Hayashi, H. Pseudokinase Tribbles1 (TRB1) negatively regulates tumor-suppressor activity of p53 through p53 deacetylation. Biol. Pharm. Bull. 2015, 38, 618–624. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; Iemura, S.I.; Natsume, T.; Miyazawa, K.; Imamura, T. Suppression of p53 activity through the cooperative action of Ski and histone deacetylase SIRT1. J. Biol. Chem. 2011, 286, 6311–6320. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.; Dai, C.; Qin, J.; Gu, W. Negative regulation of the p300-p53 interplay by DDX24. Oncogene 2016, 35, 528–536. [Google Scholar] [CrossRef]
- Furth, N.; Aylon, Y.; Oren, M. p53 shades of Hippo. Cell Death Differ. 2018, 25, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Strano, S.; Monti, O.; Pediconi, N.; Baccarini, A.; Fontemaggi, G.; Lapi, E.; Mantovani, F.; Damalas, A.; Citro, G.; Sacchi, A.; et al. The transcriptional coactivator Yes-associated protein drives p73 gene-target specificity in response to DNA damage. Mol. Cell 2005, 18, 447–459. [Google Scholar] [CrossRef]
- Escoll, M.; Gargini, R.; Cuadrado, A.; Anton, I.M.; Wandosell, F. Mutant p53 oncogenic functions in cancer stem cells are regulated by WIP through YAP/TAZ. Oncogene 2017, 36, 3515–3527. [Google Scholar] [CrossRef]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Charni, M.; Aloni-Grinstein, R.; Molchadsky, A.; Rotter, V. p53 on the crossroad between regeneration and cancer. Cell Death Differ. 2017, 24, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, Y.; Kawachi, S.; Ohkubo, T.; Nagasaka, M.; Ito, S.; Fukuura, K.; Itoh, Y.; Ohoka, N.; Morishita, D.; Hayashi, H. The CDK inhibitor p21 is a novel target gene of ATF4 and contributes to cell survival under ER stress. FEBS Lett. 2017, 591, 3682–3691. [Google Scholar] [CrossRef] [Green Version]
- Fukuura, K.; Inoue, Y.; Miyajima, C.; Watanabe, S.; Tokugawa, M.; Morishita, D.; Ohoka, N.; Komada, M.; Hayashi, H. The ubiquitin-specific protease USP17 prevents cellular senescence by stabilizing the methyltransferase SET8 and transcriptionally repressing p21. J. Biol. Chem. 2019, 294, 16429–16439. [Google Scholar] [CrossRef] [PubMed]
- Kawarada, Y.; Inoue, Y.; Kawasaki, F.; Fukuura, K.; Sato, K.; Tanaka, T.; Itoh, Y.; Hayashi, H. TGF-beta induces p53/Smads complex formation in the PAI-1 promoter to active transcription. Sci. Rep. 2016, 6, 35483. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Mora-Jensen, H.; Weniger, M.A.; Perez-Galan, P.; Wolford, C.; Hai, T.; Ron, D.; Chen, W.; Trenkle, W.; Wiestner, A.; et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2200–2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagasaka, M.; Hashimoto, R.; Inoue, Y.; Ishiuchi, K.; Matsuno, M.; Itoh, Y.; Tokugawa, M.; Ohoka, N.; Morishita, D.; Mizukami, H.; et al. Anti-tumorigenic activity of chrysin from Oroxylum indicum via non-genotoxic p53 activation through the ATM-Chk2 pathway. Molecules 2018, 23, 1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, Y.; Kitagawa, M.; Taya, Y. Phosphorylation of pRB at Ser612 by Chk1/2 leads to a complex between pRB and E2F-1 after DNA damage. EMBO J. 2007, 26, 2083–2093. [Google Scholar] [CrossRef] [Green Version]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocol to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.Y.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Lohrum, M.A.; Ludwig, R.L.; Kubbutat, M.H.; Hanlon, M.; Vousden, K.H. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 2003, 3, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Laptenko, O.; Beckerman, R.; Freulich, E.; Prives, C. p53 binding to nucleosomes within the p21 promoter in vivo leads to nucleosome loss and transcriptional activation. Proc. Natl. Acad. Sci. USA 2011, 108, 10385–10390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Huang, W.; Lei, W. Regulation and function of the TAZ transcription co-activator. Int. J. Biochem. Mol. Biol. 2011, 2, 247–256. [Google Scholar]
- Zhou, X.Z.; Lu, K.P. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer 2016, 16, 467–478. [Google Scholar] [CrossRef]
- Yaffe, M.B.; Schutkowski, M.; Shen, M.; Zhou, X.Z.; Stukenberg, P.T.; Rahfeld, J.U.; Xu, J.; Kuang, J.; Kirschner, M.W.; Fischer, G.; et al. Sequence-specific and phosphorylation-dependent proline isomerization: A potential mitotic regulatory mechanism. Science 1997, 278, 1957–1960. [Google Scholar] [CrossRef]
- Plouffe, S.W.; Lin, K.C.; Moore, J.L., III; Tan, F.E.; Ma, S.; Ye, Z.; Qiu, Y.; Ren, B.; Guan, K.L. The Hippo pathway effector proteins YAP and TAZ have both distinct and overlapping functions in the cell. J. Biol. Chem. 2018, 293, 11230–11240. [Google Scholar] [CrossRef] [Green Version]
- Morin-Kensicki, E.M.; Boone, B.N.; Howell, M.; Stonebraker, J.R.; Teed, J.; Alb, J.G.; Magnuson, T.R.; O’Neal, W.; Milgram, S.L. Defects in yolk sac vasculogenesis, chorioallantoic fusion, and embryonic axis elongation in mice with targeted disruption of Yap65. Mol. Cell. Biol. 2006, 26, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Lei, Q.Y.; Guan, K.L. The Hippo-YAP pathway: New connections between regulation of organ size and cancer. Curr. Opin. Cell Biol. 2008, 20, 638–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, M.G.; Song, H.; Shin, J.H.; Jeong, H.; Kim, H.K.; Hwang, E.S. Transcriptional coactivator with PDZ-binding motif is required to sustain testicular function on aging. Aging Cell 2017, 16, 1035–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santinon, G.; Brian, I.; Pocaterra, A.; Romani, P.; Franzolin, E.; Rampazzo, C.; Bicciato, S.; Dupont, S. dNTP metabolism links mechanical cues and YAP/TAZ to cell growth and oncogene-induced senescence. EMBO J. 2018, 37, e97780. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Saito, A.; Nagase, T. YAP/TAZ Signaling as a Molecular Link between Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 3674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef]
- Altwairgi, A.K. Statins are potential anticancerous agents. Oncol. Rep. 2015, 33, 1019–1039. [Google Scholar] [CrossRef] [Green Version]
- Mei, Z.; Liang, M.; Li, L.; Zhang, Y.; Wang, Q.; Yang, W. Effects of statins on cancer mortality and progression: A systematic review and meta-analysis of 95 cohorts including 1,111,407 individuals. Int. J. Cancer 2017, 140, 1068–1081. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyajima, C.; Kawarada, Y.; Inoue, Y.; Suzuki, C.; Mitamura, K.; Morishita, D.; Ohoka, N.; Imamura, T.; Hayashi, H. Transcriptional Coactivator TAZ Negatively Regulates Tumor Suppressor p53 Activity and Cellular Senescence. Cells 2020, 9, 171. https://doi.org/10.3390/cells9010171
Miyajima C, Kawarada Y, Inoue Y, Suzuki C, Mitamura K, Morishita D, Ohoka N, Imamura T, Hayashi H. Transcriptional Coactivator TAZ Negatively Regulates Tumor Suppressor p53 Activity and Cellular Senescence. Cells. 2020; 9(1):171. https://doi.org/10.3390/cells9010171
Chicago/Turabian StyleMiyajima, Chiharu, Yuki Kawarada, Yasumichi Inoue, Chiaki Suzuki, Kana Mitamura, Daisuke Morishita, Nobumichi Ohoka, Takeshi Imamura, and Hidetoshi Hayashi. 2020. "Transcriptional Coactivator TAZ Negatively Regulates Tumor Suppressor p53 Activity and Cellular Senescence" Cells 9, no. 1: 171. https://doi.org/10.3390/cells9010171