Protein Kinase C Alpha Cellular Distribution, Activity, and Proximity with Lamin A/C in Striated Muscle Laminopathies

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Approved Research Protocols for Human Cells and Mice Model Cells
2.2. Cell Culture and Maintenance
2.3. Cloning
2.4. Cell Transfection
2.5. Immunofluorescence
2.6. Confocal Microscopy
2.7. Cell Sorting
2.8. Protein Extraction and Western Blot
2.9. Proximity Ligation Assay (PLA)
2.10. Data Analysis
3. Results
3.1. PKC-α Localization is Disturbed in Cells Expressing Various Striated Muscle Laminopathy Mutations
3.2. PKC-α Activation Is Disturbed by Lamin A/C Mutations
3.3. PKC-α and Lamin A/C Proximity Is Disrupted by Lamin A/C Mutations
3.4. ERK 1/2 Activation Is Downregulated in Patient and Mice Model Myoblasts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Broers, J.L.; Ramaekers, F.C.; Bonne, G.; Yaou, R.B.; Hutchison, C.J. Nuclear lamins: Laminopathies and their role in premature ageing. Physiol. Rev. 2006, 86, 967–1008. [Google Scholar] [CrossRef] [PubMed]
- Tesson, F.; Saj, M.; Uvaize, M.M.; Nicolas, H.; Płoski, R.; Bilińska, Z. Lamin A/C mutations in dilated cardiomyopathy. Cardiol. J. 2014, 21, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart. J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.R.; Fain, P.R.; Sinagra, G.; Robinson, M.L.; Robertson, A.D.; Carniel, E.; Lenarda, A.D.; Bohlmeyer, T.J.; Ferguson, D.A.; Brodsky, G.L.; et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J. Am. Coll. Cardiol. 2003, 41, 771–780. [Google Scholar] [CrossRef] [Green Version]
- Pasotti, M.; Klersy, C.; Pilotto, A.; Marziliano, N.; Rapezzi, C.; Serio, A.; Mannarino, S.; Gambarin, F.; Favalli, V.; Grasso, M.; et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J. Am. Coll. Cardiol. 2008, 52, 1250–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Berlo, J.H.; de Voogt, W.G.; van der Kooi, A.J.; van Tintelen, J.P.; Bonne, G.; Yaou, R.B.; Duboc, D.; Rossenbacker, T.; Heidbüchel, H.; de Visser, M.; et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: Do lamin A/C mutations portend a high risk of sudden death? J. Mol. Med. Berl. Ger. 2005, 83, 79–83. [Google Scholar] [CrossRef]
- Hasselberg, N.E.; Haland, T.F.; Saberniak, J.; Brekke, P.H.; Berge, K.E.; Leren, T.P.; Edvardsen, T.; Haugaa, K.H. Lamin A/C cardiomyopathy: Young onset, high penetrance, and frequent need for heart transplantation. Eur. Heart J. 2018, 39, 853–860. [Google Scholar] [CrossRef]
- Colombo, M.G.; Botto, N.; Vittorini, S.; Paradossi, U.; Andreassi, M.G. Clinical utility of genetic tests for inherited hypertrophic and dilated cardiomyopathies. Cardiovasc. Ultrasound 2008, 6, 62. [Google Scholar] [CrossRef] [Green Version]
- Bonne, G.; Mercuri, E.; Muchir, A.; Urtizberea, A.; Becane, H.M.; Recan, D.; Merlini, L.; Wehnert, M.; Boor, R.; Reuner, U.; et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol. 2000, 48, 170–180. [Google Scholar] [CrossRef]
- Bonne, G.; Barletta, M.R.D.; Varnous, S.; Becane, H.M.; Hammouda, E.H.; Merlini, L.; Muntoni, F.; Greenberg, C.R.; Gary, F.; Urtizberea, J.A.; et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 1999, 21, 285–288. [Google Scholar] [CrossRef]
- Quijano-Roy, S.; Mbieleu, B.; Bönnemann, C.G.; Jeannet, P.-Y.; Colomer, J.; Clarke, N.F.; Cuisset, J.-M.; Roper, H.; De Meirleir, L.; D’Amico, A.; et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann. Neurol. 2008, 64, 177–186. [Google Scholar] [CrossRef]
- Bonne, G.; Quijano-Roy, S. Emery-Dreifuss muscular dystrophy, laminopathies, and other nuclear envelopathies. Handb. Clin. Neurol. 2013, 113, 1367–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, F.; Dabaj, I.; Mah, J.K.; Bergounioux, J.; Essid, A.; Bönnemann, C.G.; Rutkowski, A.; Bonne, G.; Quijano-Roy, S.; Wahbi, K. Cardiac manifestations of congenital LMNA-related muscular dystrophy in children: Three case reports and recommendations for care. Cardiol. Young 2017, 27, 1076–1082. [Google Scholar] [CrossRef]
- Samson, C.; Petitalot, A.; Celli, F.; Herrada, I.; Ropars, V.; Le Du, M.-H.; Nhiri, N.; Jacquet, E.; Arteni, A.-A.; Buendia, B.; et al. Structural analysis of the ternary complex between lamin A/C, BAF and emerin identifies an interface disrupted in autosomal recessive progeroid diseases. Nucleic Acids Res. 2018, 46, 10460–10473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattioli, E.; Andrenacci, D.; Garofalo, C.; Prencipe, S.; Scotlandi, K.; Remondini, D.; Gentilini, D.; Di Blasio, A.M.; Valente, S.; Scarano, E.; et al. Altered modulation of lamin A/C-HDAC2 interaction and p21 expression during oxidative stress response in HGPS. Aging Cell 2018, 17, e12824. [Google Scholar] [CrossRef] [Green Version]
- Vadrot, N.; Duband-Goulet, I.; Cabet, E.; Attanda, W.; Barateau, A.; Vicart, P.; Gerbal, F.; Briand, N.; Vigouroux, C.; Oldenburg, A.R.; et al. The p.R482W substitution in A-type lamins deregulates SREBP1 activity in Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2015, 24, 2096–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, D.N.; Zastrow, M.S.; Wilson, K.L. Direct actin binding to A- and B-type lamin tails and actin filament bundling by the lamin A tail. Nucl. Austin Tex 2010, 1, 264–272. [Google Scholar] [CrossRef]
- Lloyd, D.J.; Trembath, R.C.; Shackleton, S. A novel interaction between lamin A and SREBP1: Implications for partial lipodystrophy and other laminopathies. Hum. Mol. Genet. 2002, 11, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Bank, E.M.; Ben-Harush, K.; Wiesel-Motiuk, N.; Barkan, R.; Feinstein, N.; Lotan, O.; Medalia, O.; Gruenbaum, Y. A laminopathic mutation disrupting lamin filament assembly causes disease-like phenotypes in Caenorhabditis elegans. Mol. Biol. Cell 2011, 22, 2716–2728. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, S. Protein kinase C alpha (PKC alpha): Regulation and biological function. J. Biochem. 2002, 132, 669–675. [Google Scholar] [CrossRef]
- Martelli, A.M.; Bortul, R.; Tabellini, G.; Faenza, I.; Cappellini, A.; Bareggi, R.; Manzoli, L.; Cocco, L. Molecular characterization of protein kinase C-alpha binding to lamin A. J. Cell. Biochem. 2002, 86, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Disatnik, M.H.; Buraggi, G.; Mochly-Rosen, D. Localization of protein kinase C isozymes in cardiac myocytes. Exp. Cell Res. 1994, 210, 287–297. [Google Scholar] [CrossRef]
- Schmalz, D.; Kalkbrenner, F.; Hucho, F.; Buchner, K. Transport of protein kinase C alpha into the nucleus requires intact cytoskeleton while the transport of a protein containing a canonical nuclear localization signal does not. J. Cell Sci. 1996, 109 Pt 9, 2401–2406. [Google Scholar]
- Goodnight, J.A.; Mischak, H.; Kolch, W.; Mushinski, J.F. Immunocytochemical localization of eight protein kinase C isozymes overexpressed in NIH 3T3 fibroblasts. Isoform-specific association with microfilaments, Golgi, endoplasmic reticulum, and nuclear and cell membranes. J. Biol. Chem. 1995, 270, 9991–10001. [Google Scholar] [CrossRef] [Green Version]
- Bornancin, F.; Parker, P.J. Phosphorylation of protein kinase C-alpha on serine 657 controls the accumulation of active enzyme and contributes to its phosphatase-resistant state. J. Biol. Chem. 1997, 272, 3544–3549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazaubon, S.; Bornancin, F.; Parker, P.J. Threonine-497 is a critical site for permissive activation of protein kinase C alpha. Biochem. J. 1994, 301 Pt 2, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Parekh, D.B.; Ziegler, W.; Parker, P.J. Multiple pathways control protein kinase C phosphorylation. EMBO J. 2000, 19, 496–503. [Google Scholar] [CrossRef] [Green Version]
- Gysin, S.; Imber, R. Replacement of Ser657 of protein kinase C-alpha by alanine leads to premature down regulation after phorbol-ester-induced translocation to the membrane. Eur. J. Biochem. 1996, 240, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Keranen, L.M.; Dutil, E.M.; Newton, A.C. Protein kinase C is regulated in vivo by three functionally distinct phosphorylations. Curr. Biol. CB 1995, 5, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Braz, J.C.; Gregory, K.; Pathak, A.; Zhao, W.; Sahin, B.; Klevitsky, R.; Kimball, T.F.; Lorenz, J.N.; Nairn, A.C.; Liggett, S.B.; et al. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat. Med. 2004, 10, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Hambleton, M.; Hahn, H.; Pleger, S.T.; Kuhn, M.C.; Klevitsky, R.; Carr, A.N.; Kimball, T.F.; Hewett, T.E.; Dorn, G.W., 2nd; Koch, W.J.; et al. Pharmacological- and gene therapy-based inhibition of protein kinase Calpha/beta enhances cardiac contractility and attenuates heart failure. Circulation 2006, 114, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Pass, J.M.; Gao, J.; Jones, W.K.; Wead, W.B.; Wu, X.; Zhang, J.; Baines, C.P.; Bolli, R.; Zheng, Y.T.; Joshua, I.G.; et al. Enhanced PKC beta II translocation and PKC beta II-RACK1 interactions in PKC epsilon-induced heart failure: A role for RACK1. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H2500–H2510. [Google Scholar] [CrossRef]
- Ping, P.; Zhang, J.; Qiu, Y.; Tang, X.L.; Manchikalapudi, S.; Cao, X.; Bolli, R. Ischemic preconditioning induces selective translocation of protein kinase C isoforms epsilon and eta in the heart of conscious rabbits without subcellular redistribution of total protein kinase C activity. Circ. Res. 1997, 81, 404–414. [Google Scholar] [CrossRef]
- Bowling, N.; Walsh, R.A.; Song, G.; Estridge, T.; Sandusky, G.E.; Fouts, R.L.; Mintze, K.; Pickard, T.; Roden, R.; Bristow, M.R.; et al. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation 1999, 99, 384–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, R.; Morley, M.P.; Brandimarto, J.; Tucker, N.R.; Parsons, V.A.; Zhao, S.D.; Meder, B.; Katus, H.A.; Rühle, F.; Stoll, M.; et al. Genetic Reduction in Left Ventricular Protein Kinase C-α and Adverse Ventricular Remodeling in Human Subjects. Circ. Genomic Precis. Med. 2018, 11, e001901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouet-Benzineb, P.; Mohammadi, K.; Pérennec, J.; Poyard, M.; Nel-H, B.; Crozatier, B. Protein kinase C isoform expression in normal and failing rabbit hearts. Circ. Res. 1996, 79, 153–161. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Sentex, E.; Takeda, N.; Dhalla, N.S. Increased expression of protein kinase C isoforms in heart failure due to myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H2277–H2287. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Qian, X.; Shen, M.; Jiang, R.; Wagner, M.B.; Ding, G.; Chen, G.; Shen, B. Protein kinase C promotes cardiac fibrosis and heart failure by modulating galectin-3 expression. Biochim. Biophys. Acta 2015, 1853, 513–521. [Google Scholar] [CrossRef] [Green Version]
- Ladage, D.; Tilemann, L.; Ishikawa, K.; Correll, R.N.; Kawase, Y.; Houser, S.R.; Molkentin, J.D.; Hajjar, R.J. Inhibition of PKCα/β with ruboxistaurin antagonizes heart failure in pigs after myocardial infarction injury. Circ. Res. 2011, 109, 1396–1400. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Chen, X.; Macdonnell, S.M.; Kranias, E.G.; Lorenz, J.N.; Leitges, M.; Houser, S.R.; Molkentin, J.D. Protein kinase Calpha, but not PKCbeta or PKCgamma, regulates contractility and heart failure susceptibility: Implications for ruboxistaurin as a novel therapeutic approach. Circ. Res. 2009, 105, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Jensen, T.E.; Maarbjerg, S.J.; Rose, A.J.; Leitges, M.; Richter, E.A. Knockout of the predominant conventional PKC isoform, PKCalpha, in mouse skeletal muscle does not affect contraction-stimulated glucose uptake. Am. J. Physiol. Metab. 2009, 297, E340–E348. [Google Scholar]
- Reimann, L.; Wiese, H.; Leber, Y.; Schwäble, A.N.; Fricke, A.L.; Rohland, A.; Knapp, B.; Peikert, C.D.; Drepper, F.; van der Ven, P.F.M.; et al. Myofibrillar Z-discs Are a Protein Phosphorylation Hot Spot with Protein Kinase C (PKCα) Modulating Protein Dynamics. Mol. Cell. Proteomics MCP 2017, 16, 346–367. [Google Scholar] [CrossRef] [Green Version]
- Ivorra, C.; Kubicek, M.; González, J.M.; Sanz-González, S.M.; Alvarez-Barrientos, A.; O’Connor, J.-E.; Burke, B.; Andrés, V. A mechanism of AP-1 suppression through interaction of c-Fos with lamin A/C. Genes Dev. 2006, 20, 307–320. [Google Scholar] [CrossRef] [Green Version]
- Mauro, A.; Ciccarelli, C.; Cesaris, P.D.; Scoglio, A.; Bouche, M.; Molinaro, M.; Aquino, A.; Zani, B.M. PKCalpha-mediated ERK, JNK and p38 activation regulates the myogenic program in human rhabdomyosarcoma cells. J. Cell Sci. 2002, 115, 3587–3599. [Google Scholar] [CrossRef] [Green Version]
- Naskar, S.; Datta, K.; Mitra, A.; Pathak, K.; Datta, R.; Bansal, T.; Sarkar, S. Differential and conditional activation of PKC-isoforms dictates cardiac adaptation during physiological to pathological hypertrophy. PLoS ONE 2014, 9, e104711. [Google Scholar] [CrossRef] [Green Version]
- Schönwasser, D.C.; Marais, R.M.; Marshall, C.J.; Parker, P.J. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol. Cell. Biol. 1998, 18, 790–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soh, J.W.; Lee, E.H.; Prywes, R.; Weinstein, I.B. Novel roles of specific isoforms of protein kinase C in activation of the c-fos serum response element. Mol. Cell. Biol. 1999, 19, 1313–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soh, J.-W.; Weinstein, I.B. Roles of specific isoforms of protein kinase C in the transcriptional control of cyclin D1 and related genes. J. Biol. Chem. 2003, 278, 34709–34716. [Google Scholar] [CrossRef] [Green Version]
- Muchir, A.; Wu, W.; Choi, J.C.; Iwata, S.; Morrow, J.; Homma, S.; Worman, H.J. Abnormal p38alpha mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 2012, 21, 4325–4333. [Google Scholar] [CrossRef] [Green Version]
- Muchir, A.; Pavlidis, P.; Decostre, V.; Herron, A.J.; Arimura, T.; Bonne, G.; Worman, H.J. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Invest. 2007, 117, 1282–1293. [Google Scholar] [CrossRef]
- Muchir, A.; Kim, Y.J.; Reilly, S.A.; Wu, W.; Choi, J.C.; Worman, H.J. Inhibition of extracellular signal-regulated kinase 1/2 signaling has beneficial effects on skeletal muscle in a mouse model of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutation. Skelet. Muscle 2013, 3, 17-5040-3-17. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; Reilly, S.A.; Wu, W.; Iwata, S.; Homma, S.; Bonne, G.; Worman, H.J. Treatment with selumetinib preserves cardiac function and improves survival in cardiomyopathy caused by mutation in the lamin A/C gene. Cardiovasc. Res. 2012, 93, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muchir, A.; Shan, J.; Bonne, G.; Lehnart, S.E.; Worman, H.J. Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum. Mol. Genet. 2009, 18, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Muchir, A.; Shan, J.; Bonne, G.; Worman, H.J. Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin A/C gene. Circulation 2011, 123, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Iwata, S.; Homma, S.; Worman, H.J.; Muchir, A. Depletion of extracellular signal-regulated kinase 1 in mice with cardiomyopathy caused by lamin A/C gene mutation partially prevents pathology before isoenzyme activation. Hum. Mol. Genet. 2014, 23, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Leitges, M.; Plomann, M.; Standaert, M.L.; Bandyopadhyay, G.; Sajan, M.P.; Kanoh, Y.; Farese, R.V.; Letiges, M. Knockout of PKC alpha enhances insulin signaling through PI3K. Mol. Endocrinol. Baltim. Md 2002, 16, 847–858. [Google Scholar] [CrossRef] [Green Version]
- Braz, J.C.; Bueno, O.F.; De Windt, L.J.; Molkentin, J.D. PKC alpha regulates the hypertrophic growth of cardiomyocytes through extracellular signal-regulated kinase1/2 (ERK1/2). J. Cell Biol. 2002, 156, 905–919. [Google Scholar] [CrossRef]
- Muchir, A.; Medioni, J.; Laluc, M.; Massart, C.; Arimura, T.; van der Kooi, A.J.; Desguerre, I.; Mayer, M.; Ferrer, X.; Briault, S.; et al. Nuclear envelope alterations in fibroblasts from patients with muscular dystrophy, cardiomyopathy, and partial lipodystrophy carrying lamin A/C gene mutations. Muscle Nerve 2004, 30, 444–450. [Google Scholar] [CrossRef]
- Bertrand, A.T.; Ziaei, S.; Ehret, C.; Duchemin, H.; Mamchaoui, K.; Bigot, A.; Mayer, M.; Quijano-Roy, S.; Desguerre, I.; Lainé, J.; et al. Cellular microenvironments reveal defective mechanosensing responses and elevated YAP signaling in LMNA-mutated muscle precursors. J. Cell Sci. 2014, 127, 2873–2884. [Google Scholar] [CrossRef] [Green Version]
- Arimura, T.; Helbling-Leclerc, A.; Massart, C.; Varnous, S.; Niel, F.; Lacene, E.; Fromes, Y.; Toussaint, M.; Mura, A.M.; Keller, D.I.; et al. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet. 2005, 14, 155–169. [Google Scholar] [CrossRef]
- Bertrand, A.T.; Renou, L.; Papadopoulos, A.; Beuvin, M.; Lacène, E.; Massart, C.; Ottolenghi, C.; Decostre, V.; Maron, S.; Schlossarek, S.; et al. DelK32-lamin A/C has abnormal location and induces incomplete tissue maturation and severe metabolic defects leading to premature death. Hum. Mol. Genet. 2012, 21, 1037–1048. [Google Scholar] [CrossRef] [Green Version]
- Boudreau, E.; Labib, S.; Bertrand, A.T.; Decostre, V.; Bolongo, P.M.; Sylvius, N.; Bonne, G.; Tesson, F. Lamin A/C mutants disturb sumo1 localization and sumoylation in vitro and in vivo. PLoS ONE 2012, 7, e45918. [Google Scholar] [CrossRef] [PubMed]
- Sylvius, N.; Bilinska, Z.T.; Veinot, J.P.; Fidzianska, A.; Bolongo, P.M.; Poon, S.; McKeown, P.; Davies, R.A.; Chan, K.L.; Tang, A.S.; et al. In vivo and in vitro examination of the functional significances of novel lamin gene mutations in heart failure patients. J. Med. Genet. 2005, 42, 639–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavet, O.; Pines, J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev. Cell 2010, 18, 533–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borroni, A.P.; Emanuelli, A.; Shah, P.A.; Ilić, N.; Apel-Sarid, L.; Paolini, B.; Manikoth Ayyathan, D.; Koganti, P.; Levy-Cohen, G.; Blank, M. Smurf2 regulates stability and the autophagic-lysosomal turnover of lamin A and its disease-associated form progerin. Aging Cell 2018, 17. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, A.; Haliloglu, G.; Richard, P.; Talim, B.; Maugenre, S.; Ferreiro, A.; Guicheney, P.; Menditto, I.; Benedetti, S.; Bertini, E.; et al. Two patients with “Dropped head syndrome” due to mutations in LMNA or SEPN1 genes. Neuromuscul. Disord. NMD 2005, 15, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Vytopil, M.; Ricci, E.; Dello Russo, A.; Hanisch, F.; Neudecker, S.; Zierz, S.; Ricotti, R.; Demay, L.; Richard, P.; Wehnert, M.; et al. Frequent low penetrance mutations in the Lamin A/C gene, causing Emery Dreifuss muscular dystrophy. Neuromuscul. Disord. NMD 2002, 12, 958–963. [Google Scholar] [CrossRef]
- Fidzianska, A.; Bilinska, Z.T.; Tesson, F.; Wagner, T.; Walski, M.; Grzybowski, J.; Ruzyllo, W.; Hausmanowa-Petrusewicz, I. Obliteration of cardiomyocyte nuclear architecture in a patient with LMNA gene mutation. J. Neurol. Sci. 2008, 271, 91–96. [Google Scholar] [CrossRef]
- Bilinska, Z.T.; Sylvius, N.; Grzybowski, J.; Fidzianska, A.; Michalak, E.; Walczak, E.; Walski, M.; Bieganowska, K.; Szymaniak, E.; Kusmierczyk-Droszcz, B.; et al. Dilated cardiomyopathy caused by LMNA mutations. Clinical and morphological studies. Kardiol. Pol. 2006, 64, 812–819; discussion 820–821. [Google Scholar]
- Meng, Q.; Li, B.X.; Xiao, X. Toward Developing Chemical Modulators of Hsp60 as Potential Therapeutics. Front. Mol. Biosci. 2018, 5, 35. [Google Scholar] [CrossRef] [Green Version]
- Itoh, H.; Komatsuda, A.; Ohtani, H.; Wakui, H.; Imai, H.; Sawada, K.-I.; Otaka, M.; Ogura, M.; Suzuki, A.; Hamada, F. Mammalian HSP60 is quickly sorted into the mitochondria under conditions of dehydration. Eur. J. Biochem. 2002, 269, 5931–5938. [Google Scholar] [CrossRef] [PubMed]
- Deniset, J.F.; Hedley, T.E.; Hlaváčková, M.; Chahine, M.N.; Dibrov, E.; O’Hara, K.; Maddaford, G.G.; Nelson, D.; Maddaford, T.G.; Fandrich, R.; et al. Heat shock protein 60 involvement in vascular smooth muscle cell proliferation. Cell. Signal. 2018, 47, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Itoh, H.; Kobayashi, R.; Wakui, H.; Komatsuda, A.; Ohtani, H.; Miura, A.B.; Otaka, M.; Masamune, O.; Andoh, H.; Koyama, K. Mammalian 60-kDa stress protein (chaperonin homolog). Identification, biochemical properties, and localization. J. Biol. Chem. 1995, 270, 13429–13435. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.S. Proximity Ligation Assay (PLA). Curr. Protoc. Immunol. 2018, 123, e58. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Arneja, A.S.; Dhalla, N.S. Alterations in protein kinase A and protein kinase C levels in heart failure due to genetic cardiomyopathy. Can. J. Cardiol. 1999, 15, 683–690. [Google Scholar]
- Gigli, M.; Stolfo, D.; Merlo, M.; Barbati, G.; Ramani, F.; Brun, F.; Pinamonti, B.; Sinagra, G. Insights into mildly dilated cardiomyopathy: Temporal evolution and long-term prognosis. Eur. J. Heart Fail. 2017, 19, 531–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, J.M.; Navarro-Puche, A.; Casar, B.; Crespo, P.; Andrés, V. Fast regulation of AP-1 activity through interaction of lamin A/C, ERK1/2, and c-Fos at the nuclear envelope. J. Cell Biol. 2008, 183, 653–666. [Google Scholar] [CrossRef] [Green Version]
- Myant, K.; Qiao, X.; Halonen, T.; Come, C.; Laine, A.; Janghorban, M.; Partanen, J.I.; Cassidy, J.; Ogg, E.-L.; Cammareri, P.; et al. Serine 62-Phosphorylated MYC Associates with Nuclear Lamins and Its Regulation by CIP2A Is Essential for Regenerative Proliferation. Cell Rep. 2015, 12, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, J.; Calvo, F.; González, J.M.; Casar, B.; Andrés, V.; Crespo, P. ERK1/2 MAP kinases promote cell cycle entry by rapid, kinase-independent disruption of retinoblastoma-lamin A complexes. J. Cell Biol. 2010, 191, 967–979. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Domínguez, D.; Epifano, C.; de Miguel, F.; Castaño, A.G.; Vilaplana-Martí, B.; Martín, A.; Amarilla-Quintana, S.; Bertrand, A.T.; Bonne, G.; Ramón-Azcón, J.; et al. Consequences of Lmna Exon 4 Mutations in Myoblast Function. Cells 2020, 9, 1286. [Google Scholar] [CrossRef]
- Zwerger, M.; Jaalouk, D.E.; Lombardi, M.L.; Isermann, P.; Mauermann, M.; Dialynas, G.; Herrmann, H.; Wallrath, L.L.; Lammerding, J. Myopathic lamin mutations impair nuclear stability in cells and tissue and disrupt nucleo-cytoskeletal coupling. Hum. Mol. Genet. 2013, 22, 2335–2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.C.; Wu, W.; Muchir, A.; Iwata, S.; Homma, S.; Worman, H.J. Dual specificity phosphatase 4 mediates cardiomyopathy caused by lamin A/C (LMNA) gene mutation. J. Biol. Chem. 2012, 287, 40513–40524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frémin, C.; Saba-El-Leil, M.K.; Lévesque, K.; Ang, S.-L.; Meloche, S. Functional Redundancy of ERK1 and ERK2 MAP Kinases during Development. Cell Rep. 2015, 12, 913–921. [Google Scholar] [CrossRef] [Green Version]
- Krens, S.F.G.; He, S.; Spaink, H.P.; Snaar-Jagalska, B.E. Characterization and expression patterns of the MAPK family in zebrafish. Gene Expr. Patterns GEP 2006, 6, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Krens, S.F.G.; He, S.; Lamers, G.E.M.; Meijer, A.H.; Bakkers, J.; Schmidt, T.; Spaink, H.P.; Snaar-Jagalska, B.E. Distinct functions for ERK1 and ERK2 in cell migration processes during zebrafish gastrulation. Dev. Biol. 2008, 319, 370–383. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
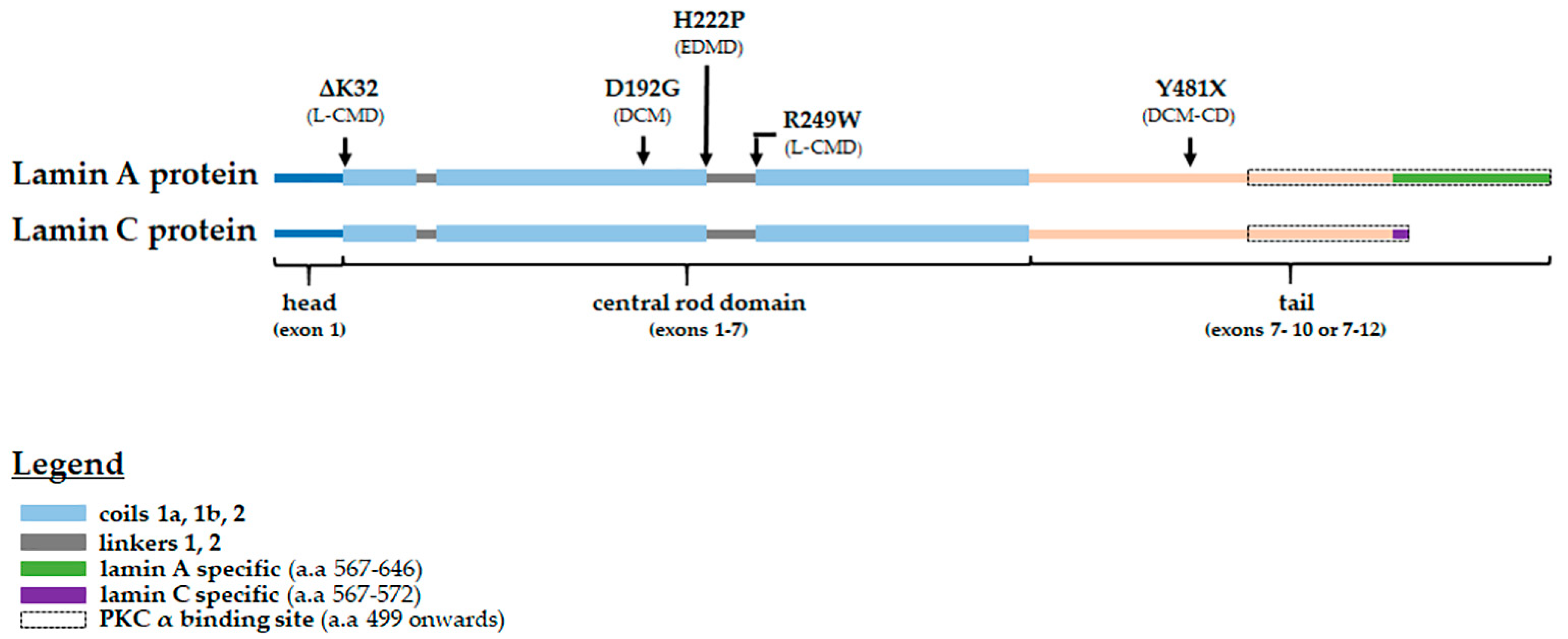
| Variant | Phenotype (s) | Reference (s) |
|---|---|---|
| p.delK32 | L-CMD *, severe EDMD | [66,67] |
| p.D192G | severe DCM | [63,68] |
| p.H222P | EDMD with arrhythmia | [9] |
| p.R249W | L-CMD | [11] |
| p.Y481X | DCM-CD | [63,69] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicolas, H.A.; Bertrand, A.T.; Labib, S.; Mohamed-Uvaize, M.; Bolongo, P.M.; Wu, W.Y.; Bilińska, Z.T.; Bonne, G.; Akimenko, M.-A.; Tesson, F. Protein Kinase C Alpha Cellular Distribution, Activity, and Proximity with Lamin A/C in Striated Muscle Laminopathies. Cells 2020, 9, 2388. https://doi.org/10.3390/cells9112388
Nicolas HA, Bertrand AT, Labib S, Mohamed-Uvaize M, Bolongo PM, Wu WY, Bilińska ZT, Bonne G, Akimenko M-A, Tesson F. Protein Kinase C Alpha Cellular Distribution, Activity, and Proximity with Lamin A/C in Striated Muscle Laminopathies. Cells. 2020; 9(11):2388. https://doi.org/10.3390/cells9112388
Chicago/Turabian StyleNicolas, Hannah A., Anne T. Bertrand, Sarah Labib, Musfira Mohamed-Uvaize, Pierrette M. Bolongo, Wen Yu Wu, Zofia T. Bilińska, Gisèle Bonne, Marie-Andrée Akimenko, and Frédérique Tesson. 2020. "Protein Kinase C Alpha Cellular Distribution, Activity, and Proximity with Lamin A/C in Striated Muscle Laminopathies" Cells 9, no. 11: 2388. https://doi.org/10.3390/cells9112388