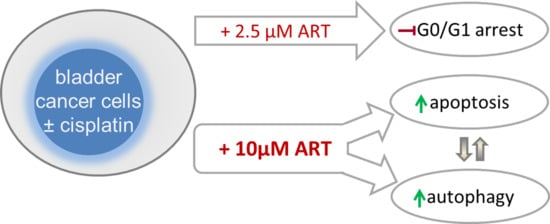
Artesunate Impairs Growth in Cisplatin-Resistant Bladder Cancer Cells by Cell Cycle Arrest, Apoptosis and Autophagy Induction


 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Resistance Induction and Drug Treatment
2.3. Tumor Cell Growth
2.4. Proliferation
2.5. Cell Cycle Phase Distribution
2.6. Apoptosis and Necrosis
2.7. Evaluation of Mitochondrial Respiration and Anaerobic Glycolytic Activity
2.8. Ferroptosis
2.9. Expression of Cell Cycle and Cell Death Regulating Proteins
2.10. Statistics
3. Results
3.1. ART Induced Significant Growth Inhibition of Parental and Cisplatin-Resistant Cells
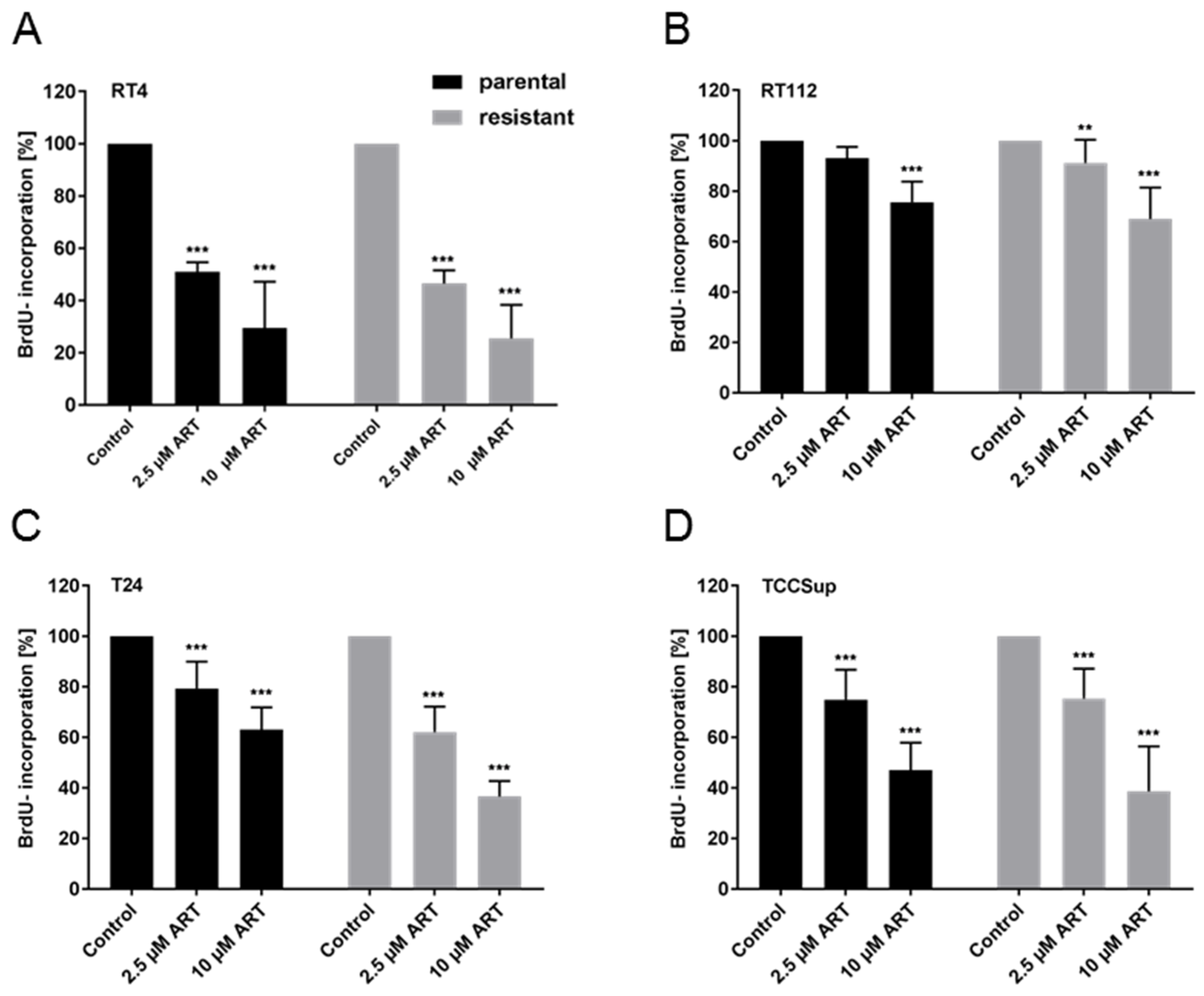
3.2. Proliferation of BCa Cells Significantly Decreased upon ART Application
3.3. ART Resulted in Cell Cycle Arrest and Elevation of Apoptotic Events
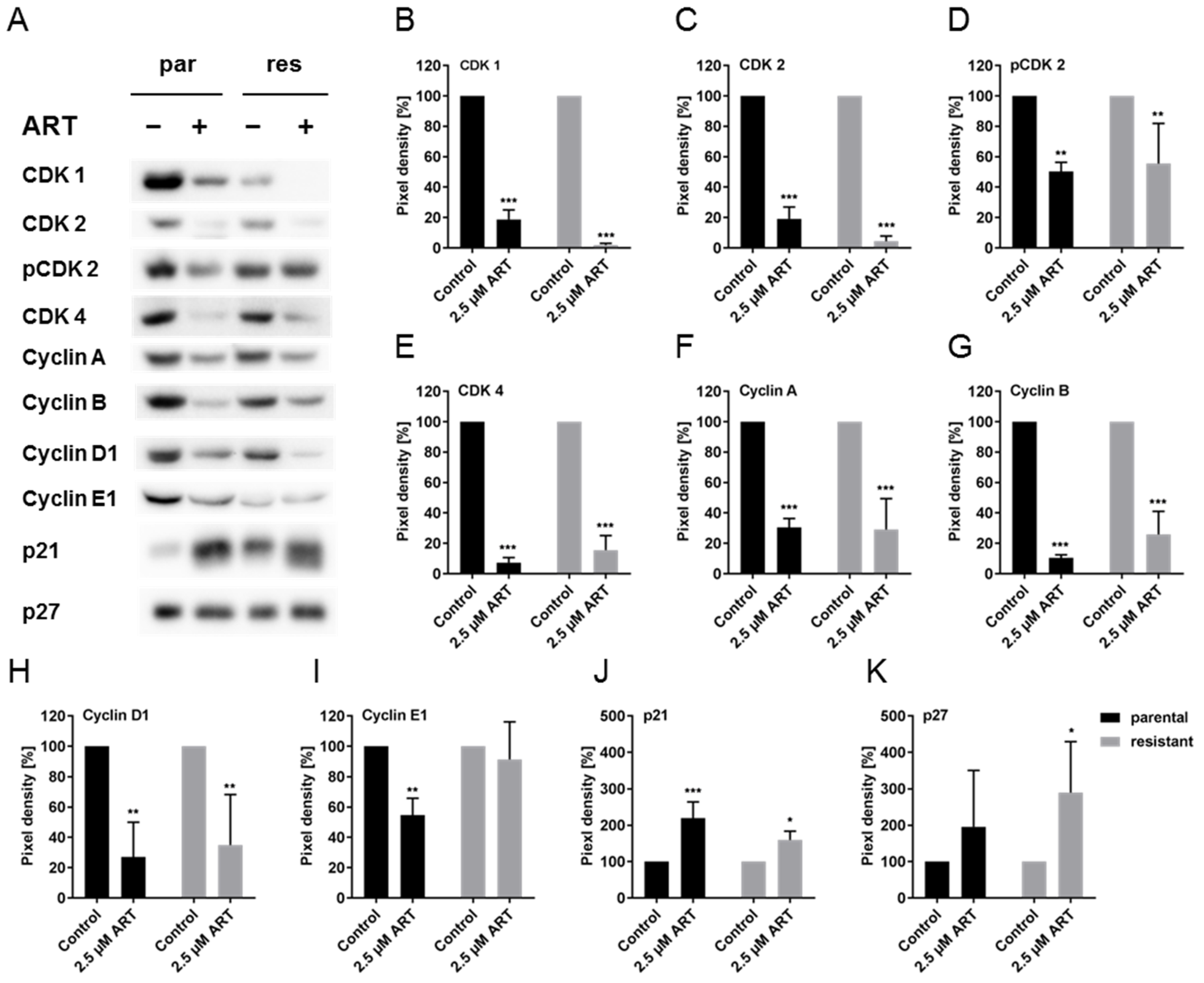
3.4. ART Modulated Cell Cycle Regulating Proteins
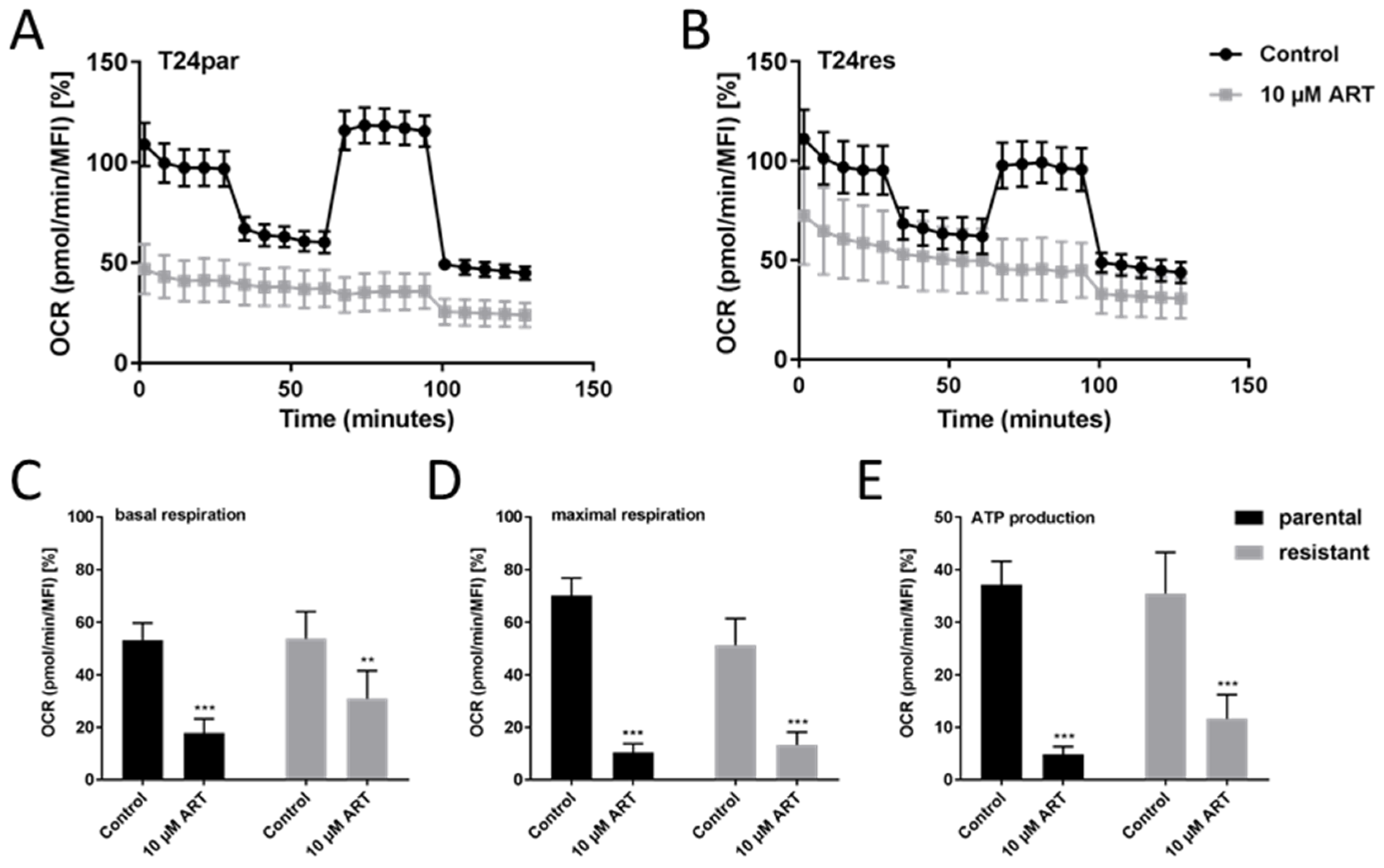
3.5. ART Disrupted Mitochondrial Respiration
3.6. ART Inhibited DNA Damage Repair and Modulated Cell Death-Associated Proteins
3.7. ART Did Not Induce Ferroptosis in BCa Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Retz, M.; Gschwend, J.E.; Maisch, P. [Short version of the German S3 guideline for bladder cancer]. Urologe A 2016, 55, 1173–1187. [Google Scholar] [CrossRef] [PubMed]
- Witjes, J.A.; Bruins, H.M.; Cathomas, R.; Comperat, E.M.; Cowan, N.C.; Gakis, G.; Hernandez, V.; Linares Espinos, E.; Lorch, A.; Neuzillet, Y.; et al. European Association of Urology Guidelines on Muscle-invasive and Metastatic Bladder Cancer: Summary of the 2020 Guidelines. Eur. Urol. 2020. [Google Scholar] [CrossRef] [PubMed]
- World Health Oiganization. Bladder. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/30-Bladder-fact-sheet.pdf (accessed on 9 December 2020).
- Torgovnick, A.; Schumacher, B. DNA repair mechanisms in cancer development and therapy. Front. Genet. 2015, 6, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [Green Version]
- Giacomini, I.; Ragazzi, E.; Pasut, G.; Montopoli, M. The Pentose Phosphate Pathway and Its Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2020, 21, 937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadal, R.; Bellmunt, J. Management of metastatic bladder cancer. Cancer Treat. Rev. 2019, 76, 10–21. [Google Scholar] [CrossRef]
- Liu, S.T.; Hui, G.; Mathis, C.; Chamie, K.; Pantuck, A.J.; Drakaki, A. The Current Status and Future Role of the Phosphoinositide 3 Kinase/AKT Signaling Pathway in Urothelial Cancer: An Old Pathway in the New Immunotherapy Era. Clin. Genitourin Cancer 2018, 16, e269–e276. [Google Scholar] [CrossRef]
- Luo, J.; Zhu, W.; Tang, Y.; Cao, H.; Zhou, Y.; Ji, R.; Zhou, X.; Lu, Z.; Yang, H.; Zhang, S.; et al. Artemisinin derivative artesunate induces radiosensitivity in cervical cancer cells in vitro and in vivo. Radiat. Oncol. 2014, 9, 84. [Google Scholar] [CrossRef] [Green Version]
- Fei, Z.; Gu, W.; Xie, R.; Su, H.; Jiang, Y. Artesunate enhances radiosensitivity of esophageal cancer cells by inhibiting the repair of DNA damage. J. Pharmacol. Sci. 2018, 138, 131–137. [Google Scholar] [CrossRef]
- Wang, B.; Hou, D.; Liu, Q.; Wu, T.; Guo, H.; Zhang, X.; Zou, Y.; Liu, Z.; Liu, J.; Wei, J.; et al. Artesunate sensitizes ovarian cancer cells to cisplatin by downregulating RAD51. Cancer Biol. Ther. 2015, 16, 1548–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdelle, N.; Nikolova, T.; Quiros, S.; Efferth, T.; Kaina, B. Artesunate induces oxidative DNA damage, sustained DNA double-strand breaks, and the ATM/ATR damage response in cancer cells. Mol. Cancer Ther. 2011, 10, 2224–2233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelis, M.; Kleinschmidt, M.C.; Barth, S.; Rothweiler, F.; Geiler, J.; Breitling, R.; Mayer, B.; Deubzer, H.; Witt, O.; Kreuter, J.; et al. Anti-cancer effects of artesunate in a panel of chemoresistant neuroblastoma cell lines. Biochem. Pharmacol. 2010, 79, 130–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, J.L.; Kim, E.H.; Jang, H.J.; Park, J.Y.; Shin, D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 2016, 381, 96–103. [Google Scholar] [CrossRef]
- Roh, J.L.; Kim, E.H.; Jang, H.; Shin, D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017, 11, 254–262. [Google Scholar] [CrossRef]
- Cumming, J.N.; Ploypradith, P.; Posner, G.H. Antimalarial activity of artemisinin (qinghaosu) and related trioxanes: Mechanism(s) of action. Adv. Pharmacol. 1997, 37, 253–297. [Google Scholar] [CrossRef]
- O’Neill, P.M.; Posner, G.H. A medicinal chemistry perspective on artemisinin and related endoperoxides. J. Med. Chem. 2004, 47, 2945–2964. [Google Scholar] [CrossRef]
- Aulbert, E.; Disselhoff, W.; Sorje, H.; Schulz, E.; Gericke, D. Lysosomal accumulation of 67Ga--transferrin in malignant tumors in relation to their growth rate. Eur. J. Cancer 1980, 16, 1217–1232. [Google Scholar] [CrossRef]
- Shterman, N.; Kupfer, B.; Moroz, C. Comparison of transferrin receptors, iron content and isoferritin profile in normal and malignant human breast cell lines. Pathobiology 1991, 59, 19–25. [Google Scholar] [CrossRef]
- Gatter, K.C.; Brown, G.; Trowbridge, I.S.; Woolston, R.E.; Mason, D.Y. Transferrin receptors in human tissues: Their distribution and possible clinical relevance. J. Clin. Pathol. 1983, 36, 539–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Judd, W.; Poodry, C.A.; Strominger, J.L. Novel surface antigen expressed on dividing cells but absent from nondividing cells. J. Exp. Med. 1980, 152, 1430–1435. [Google Scholar] [CrossRef] [PubMed]
- Trowbridge, I.S.; Omary, M.B. Human cell surface glycoprotein related to cell proliferation is the receptor for transferrin. Proc. Natl. Acad. Sci. USA 1981, 78, 3039–3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, R.; Delia, D.; Schneider, C.; Newman, R.; Kemshead, J.; Greaves, M. Ubiquitous cell-surface glycoprotein on tumor cells is proliferation-associated receptor for transferrin. Proc. Natl. Acad. Sci. USA 1981, 78, 4515–4519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efferth, T. From ancient herb to modern drug: Artemisia annua and artemisinin for cancer therapy. Semin. Cancer Biol. 2017, 46, 65–83. [Google Scholar] [CrossRef]
- Efferth, T.; Benakis, A.; Romero, M.R.; Tomicic, M.; Rauh, R.; Steinbach, D.; Hafer, R.; Stamminger, T.; Oesch, F.; Kaina, B.; et al. Enhancement of cytotoxicity of artemisinins toward cancer cells by ferrous iron. Free. Radic. Biol. Med. 2004, 37, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Kelter, G.; Steinbach, D.; Konkimalla, V.B.; Tahara, T.; Taketani, S.; Fiebig, H.H.; Efferth, T. Role of transferrin receptor and the ABC transporters ABCB6 and ABCB7 for resistance and differentiation of tumor cells towards artesunate. PLoS ONE 2007, 2, e798. [Google Scholar] [CrossRef] [Green Version]
- Saeed, M.E.M.; Breuer, E.; Hegazy, M.F.; Efferth, T. Retrospective study of small pet tumors treated with Artemisia annua and iron. Int. J. Oncol. 2020, 56, 123–138. [Google Scholar] [CrossRef]
- Efferth, T.; Giaisi, M.; Merling, A.; Krammer, P.H.; Li-Weber, M. Artesunate induces ROS-mediated apoptosis in doxorubicin-resistant T leukemia cells. PLoS ONE 2007, 2, e693. [Google Scholar] [CrossRef] [Green Version]
- Ooko, E.; Saeed, M.E.; Kadioglu, O.; Sarvi, S.; Colak, M.; Elmasaoudi, K.; Janah, R.; Greten, H.J.; Efferth, T. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine 2015, 22, 1045–1054. [Google Scholar] [CrossRef]
- Efferth, T.; Rucker, G.; Falkenberg, M.; Manns, D.; Olbrich, A.; Fabry, U.; Osieka, R. Detection of apoptosis in KG-1a leukemic cells treated with investigational drugs. Arzneimittelforschung 1996, 46, 196–200. [Google Scholar]
- Jiang, F.; Zhou, J.Y.; Zhang, D.; Liu, M.H.; Chen, Y.G. Artesunate induces apoptosis and autophagy in HCT116 colon cancer cells, and autophagy inhibition enhances the artesunateinduced apoptosis. Int. J. Mol. Med. 2018, 42, 1295–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenshields, A.L.; Fernando, W.; Hoskin, D.W. The anti-malarial drug artesunate causes cell cycle arrest and apoptosis of triple-negative MDA-MB-468 and HER2-enriched SK-BR-3 breast cancer cells. Exp. Mol. Pathol. 2019, 107, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Zuo, W.; Wang, Z.Z.; Xue, J. Artesunate induces apoptosis of bladder cancer cells by miR-16 regulation of COX-2 expression. Int. J. Mol. Sci. 2014, 15, 14298–14312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Bu, S.; Sun, J.; Guo, Y.; Lai, D. Artemisinin derivatives inhibit epithelial ovarian cancer cells via autophagy-mediated cell cycle arrest. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 1227–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Q.; Chen, H.; Xiong, G.; Jiao, R.; Liu, Y.; Li, X.; Sun, Y.; Wang, J.; Yan, L. Artesunate protects against surgery-induced knee arthrofibrosis by activating Beclin-1-mediated autophagy via inhibition of mTOR signaling. Eur. J. Pharmacol. 2019, 854, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Wass, M.N.; Cinatl, J. Drug-adapted cancer cell lines as preclinical models of acquired resistance. Cancer Drug. Resist. 2019, 2, 447–456. [Google Scholar] [CrossRef] [Green Version]
- Michaelis, M.; Rothweiler, F.; Barth, S.; Cinatl, J.; van Rikxoort, M.; Loschmann, N.; Voges, Y.; Breitling, R.; von Deimling, A.; Rodel, F.; et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011, 2, e243. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, X.; Wang, L.; Wong, Y.K.; Lee, Y.M.; Zhou, C.; Wu, G.; Zhao, T.; Yang, L.; Lu, L.; et al. Artesunate-induced mitophagy alters cellular redox status. Redox Biol. 2018, 19, 263–273. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Davis, T.M.; Phuong, H.L.; Ilett, K.F.; Hung, N.C.; Batty, K.T.; Phuong, V.D.; Powell, S.M.; Thien, H.V.; Binh, T.Q. Pharmacokinetics and pharmacodynamics of intravenous artesunate in severe falciparum malaria. Antimicrob. Agents Chemother. 2001, 45, 181–186. [Google Scholar] [CrossRef] [Green Version]
- Vatsveen, T.K.; Myhre, M.R.; Steen, C.B.; Walchli, S.; Lingjaerde, O.C.; Bai, B.; Dillard, P.; Theodossiou, T.A.; Holien, T.; Sundan, A.; et al. Artesunate shows potent anti-tumor activity in B-cell lymphoma. J. Hematol. Oncol. 2018, 11, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Xu, K.; Pian, G.; Sun, S. Artesunate and sorafenib: Combinatorial inhibition of liver cancer cell growth. Oncol. Lett. 2019, 18, 4735–4743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Shou, L.M.; Lin, F.; Duan, W.M.; Wu, M.Y.; Xie, X.; Xie, Y.F.; Li, W.; Tao, M. Artesunate induces G2/M cell cycle arrest through autophagy induction in breast cancer cells. Anticancer Drugs. 2014, 25, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Yang, L.; Hu, H.; Ke, Y. Artesunate targets oral tongue squamous cell carcinoma via mitochondrial dysfunction-dependent oxidative damage and Akt/AMPK/mTOR inhibition. J. Bioenerg. Biomembr. 2020, 52, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Trimble, C.L.; Levinson, K.; Maldonado, L.; Donovan, M.J.; Clark, K.T.; Fu, J.; Shay, M.E.; Sauter, M.E.; Sanders, S.A.; Frantz, P.S.; et al. A first-in-human proof-of-concept trial of intravaginal artesunate to treat cervical intraepithelial neoplasia 2/3 (CIN2/3). Gynecol. Oncol. 2020, 157, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, X.L.; Zhang, G.H.; Gao, Y.F. Artesunate promotes Th1 differentiation from CD4+ T cells to enhance cell apoptosis in ovarian cancer via miR-142. Braz. J. Med. Biol. Res. 2019, 52, e7992. [Google Scholar] [CrossRef] [PubMed]
- Greenshields, A.L.; Shepherd, T.G.; Hoskin, D.W. Contribution of reactive oxygen species to ovarian cancer cell growth arrest and killing by the anti-malarial drug artesunate. Mol. Carcinog. 2017, 56, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zuo, L.F.; Zuo, J.; Wang, J. Artesunate induces apoptosis and inhibits growth of Eca109 and Ec9706 human esophageal cancer cell lines in vitro and in vivo. Mol. Med. Rep. 2015, 12, 1465–1472. [Google Scholar] [CrossRef]
- Kumar, V.L.; Verma, S.; Das, P. Artesunate suppresses inflammation and oxidative stress in a rat model of colorectal cancer. Drug. Dev. Res. 2019, 80, 1089–1097. [Google Scholar] [CrossRef]
- Verma, S.; Das, P.; Kumar, V.L. Chemoprevention by artesunate in a preclinical model of colorectal cancer involves down regulation of beta-catenin, suppression of angiogenesis, cellular proliferation and induction of apoptosis. Chem. Biol. Interact. 2017, 278, 84–91. [Google Scholar] [CrossRef]
- Li, P.C.; Lam, E.; Roos, W.P.; Zdzienicka, M.Z.; Kaina, B.; Efferth, T. Artesunate derived from traditional Chinese medicine induces DNA damage and repair. Cancer Res. 2008, 68, 4347–4351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efferth, T.; Sauerbrey, A.; Olbrich, A.; Gebhart, E.; Rauch, P.; Weber, H.O.; Hengstler, J.G.; Halatsch, M.E.; Volm, M.; Tew, K.D.; et al. Molecular modes of action of artesunate in tumor cell lines. Mol. Pharmacol. 2003, 64, 382–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamacher-Brady, A.; Stein, H.A.; Turschner, S.; Toegel, I.; Mora, R.; Jennewein, N.; Efferth, T.; Eils, R.; Brady, N.R. Artesunate activates mitochondrial apoptosis in breast cancer cells via iron-catalyzed lysosomal reactive oxygen species production. J. Biol. Chem. 2011, 286, 6587–6601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, Y.; Liu, Y.; Zheng, H.; Zheng, L.; Liu, W.; Wu, J.; Ou, R.; Zhang, G.; Li, F.; Hu, M.; et al. Artemisinin and its derivatives can significantly inhibit lung tumorigenesis and tumor metastasis through Wnt/beta-catenin signaling. Oncotarget 2016, 7, 31413–31428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tin, A.S.; Sundar, S.N.; Tran, K.Q.; Park, A.H.; Poindexter, K.M.; Firestone, G.L. Antiproliferative effects of artemisinin on human breast cancer cells requires the downregulated expression of the E2F1 transcription factor and loss of E2F1-target cell cycle genes. Anticancer Drugs 2012, 23, 370–379. [Google Scholar] [CrossRef]
- Zhang, H.T.; Wang, Y.L.; Zhang, J.; Zhang, Q.X. Artemisinin inhibits gastric cancer cell proliferation through upregulation of p53. Tumour. Biol. 2014, 35, 1403–1409. [Google Scholar] [CrossRef]
- Chen, M.; Huang, J.; Zhu, Z.; Zhang, J.; Li, K. Systematic review and meta-analysis of tumor biomarkers in predicting prognosis in esophageal cancer. BMC Cancer 2013, 13, 539. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Cai, J.; Yang, Q.; Ding, H.; Wu, L.; Li, T.; Wang, Z. Prognostic significance of several biomarkers in epithelial ovarian cancer: A meta-analysis of published studies. J. Cancer Res. Clin. Oncol. 2013, 139, 1257–1277. [Google Scholar] [CrossRef]
- Tang, K.; Wang, C.; Chen, Z.; Xu, H.; Ye, Z. Clinicopathologic and prognostic significance of p21 (Cip1/Waf1) expression in bladder cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 4999–5007. [Google Scholar]
- Wang, C.; Chen, Z.; Ge, Q.; Hu, J.; Li, F.; Hu, J.; Xu, H.; Ye, Z.; Li, L.C. Up-regulation of p21(WAF1/CIP1) by miRNAs and its implications in bladder cancer cells. FEBS Lett. 2014, 588, 4654–4664. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Chen, Y.; Wang, F.; Wu, H.; Zhang, Y.; Liu, J.; Cai, Y.; Huang, S.; He, N.; Hu, Z.; et al. Artesunate induces autophagy dependent apoptosis through upregulating ROS and activating AMPK-mTOR-ULK1 axis in human bladder cancer cells. Chem. Biol. Interact. 2020, 331, 109273. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, L.; Wang, J.; Chen, Y. Inhibitory Effect of Artesunate on Growth and Apoptosis of Gastric Cancer Cells. Arch. Med. Res. 2017, 48, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Parsons, M.J.; Llambi, F.; Bouchier-Hayes, L.; Connell, S.; Munoz-Pinedo, C.; Green, D.R. Resistance to caspase-independent cell death requires persistence of intact mitochondria. Dev. Cell 2010, 18, 802–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, S.Y.; Najjar, T.A.; Alashari, M. Role of non-selective adenosine receptor blockade and phosphodiesterase inhibition in cisplatin-induced nephrogonadal toxicity in rats. Clin. Exp. Pharmacol. Physiol. 2004, 31, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ni, W.; Deng, Z.; Liu, M.; She, L.; Xie, Q. Targeting nasopharyngeal carcinoma by artesunate through inhibiting Akt/mTOR and inducing oxidative stress. Fundam. Clin. Pharmacol. 2017, 31, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, J.; Chen, Q.; Han, S.; Shao, H.; Chen, P.; Jin, Q.; Yang, M.; Shangguan, F.; Fei, M.; et al. Artemisinin suppresses hepatocellular carcinoma cell growth, migration and invasion by targeting cellular bioenergetics and Hippo-YAP signaling. Arch. Toxicol. 2019, 93, 3367–3383. [Google Scholar] [CrossRef]
- Lartigue, L.; Kushnareva, Y.; Seong, Y.; Lin, H.; Faustin, B.; Newmeyer, D.D. Caspase-independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Mol. Biol. Cell 2009, 20, 4871–4884. [Google Scholar] [CrossRef] [Green Version]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015, 2, 517–532. [Google Scholar] [CrossRef] [Green Version]
- Pinto-Leite, R.; Arantes-Rodrigues, R.; Ferreira, R.; Palmeira, C.; Colaco, A.; Moreira da Silva, V.; Oliveira, P.; Lara Santos, L. Temsirolimus improves cytotoxic efficacy of cisplatin and gemcitabine against urinary bladder cancer cell lines. Urol. Oncol. 2014, 32, 41.e11–e12. [Google Scholar] [CrossRef]
- Chen, P.; Huang, H.P.; Wang, Y.; Jin, J.; Long, W.G.; Chen, K.; Zhao, X.H.; Chen, C.G.; Li, J. Curcumin overcome primary gefitinib resistance in non-small-cell lung cancer cells through inducing autophagy-related cell death. J. Exp. Clin. Cancer Res. 2019, 38, 254. [Google Scholar] [CrossRef] [Green Version]

 G2/,
G2/,  S,
S,  GO/G1) of parental and cisplatin-resistant T24 (A) and TCCSup (B) cells after 48 h (TCCSup) or 72 h (T24) exposure to ART. Percent of apoptotic events in parental and resistant T24 (C) and TCCSup (D) cells after 72 h treatment with ART (2.5 and 10 µM); comparison is to untreated controls. Error bars indicate standard deviation (SD). Significant difference to untreated control: * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. n = 4.
G2/, S, GO/G1) of parental and cisplatin-resistant T24 (A) and TCCSup (B) cells after 48 h (TCCSup) or 72 h (T24) exposure to ART. Percent of apoptotic events in parental and resistant T24 (C) and TCCSup (D) cells after 72 h treatment with ART (2.5 and 10 µM); comparison is to untreated controls. Error bars indicate standard deviation (SD). Significant difference to untreated control: * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. n = 4.
GO/G1) of parental and cisplatin-resistant T24 (A) and TCCSup (B) cells after 48 h (TCCSup) or 72 h (T24) exposure to ART. Percent of apoptotic events in parental and resistant T24 (C) and TCCSup (D) cells after 72 h treatment with ART (2.5 and 10 µM); comparison is to untreated controls. Error bars indicate standard deviation (SD). Significant difference to untreated control: * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. n = 4.
G2/, S, GO/G1) of parental and cisplatin-resistant T24 (A) and TCCSup (B) cells after 48 h (TCCSup) or 72 h (T24) exposure to ART. Percent of apoptotic events in parental and resistant T24 (C) and TCCSup (D) cells after 72 h treatment with ART (2.5 and 10 µM); comparison is to untreated controls. Error bars indicate standard deviation (SD). Significant difference to untreated control: * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. n = 4.


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, F.; Vakhrusheva, O.; Markowitsch, S.D.; Slade, K.S.; Tsaur, I.; Cinatl, J., Jr.; Michaelis, M.; Efferth, T.; Haferkamp, A.; Juengel, E. Artesunate Impairs Growth in Cisplatin-Resistant Bladder Cancer Cells by Cell Cycle Arrest, Apoptosis and Autophagy Induction. Cells 2020, 9, 2643. https://doi.org/10.3390/cells9122643
Zhao F, Vakhrusheva O, Markowitsch SD, Slade KS, Tsaur I, Cinatl J Jr., Michaelis M, Efferth T, Haferkamp A, Juengel E. Artesunate Impairs Growth in Cisplatin-Resistant Bladder Cancer Cells by Cell Cycle Arrest, Apoptosis and Autophagy Induction. Cells. 2020; 9(12):2643. https://doi.org/10.3390/cells9122643
Chicago/Turabian StyleZhao, Fuguang, Olesya Vakhrusheva, Sascha D. Markowitsch, Kimberly S. Slade, Igor Tsaur, Jindrich Cinatl, Jr., Martin Michaelis, Thomas Efferth, Axel Haferkamp, and Eva Juengel. 2020. "Artesunate Impairs Growth in Cisplatin-Resistant Bladder Cancer Cells by Cell Cycle Arrest, Apoptosis and Autophagy Induction" Cells 9, no. 12: 2643. https://doi.org/10.3390/cells9122643