Unraveling LMNA Mutations in Metabolic Syndrome: Cellular Phenotype and Clinical Pitfalls

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Patients
2.2. Plasmids and siRNAs
2.3. Cell Culture and Cell Transfection
2.4. Senescence Evaluation
2.5. RNA Sequencing and RT-qPCR
2.6. Statistical Analysis
3. Results
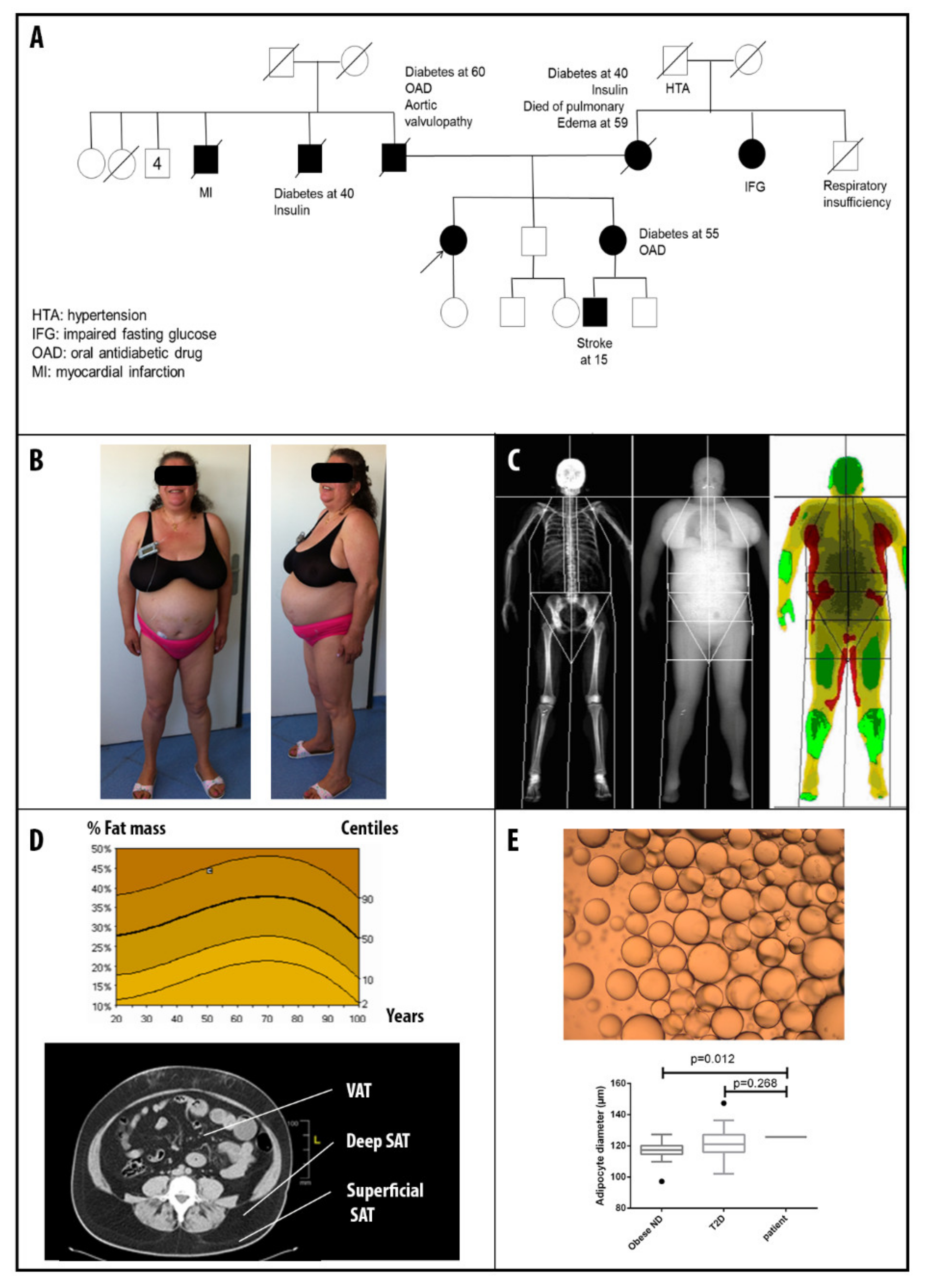
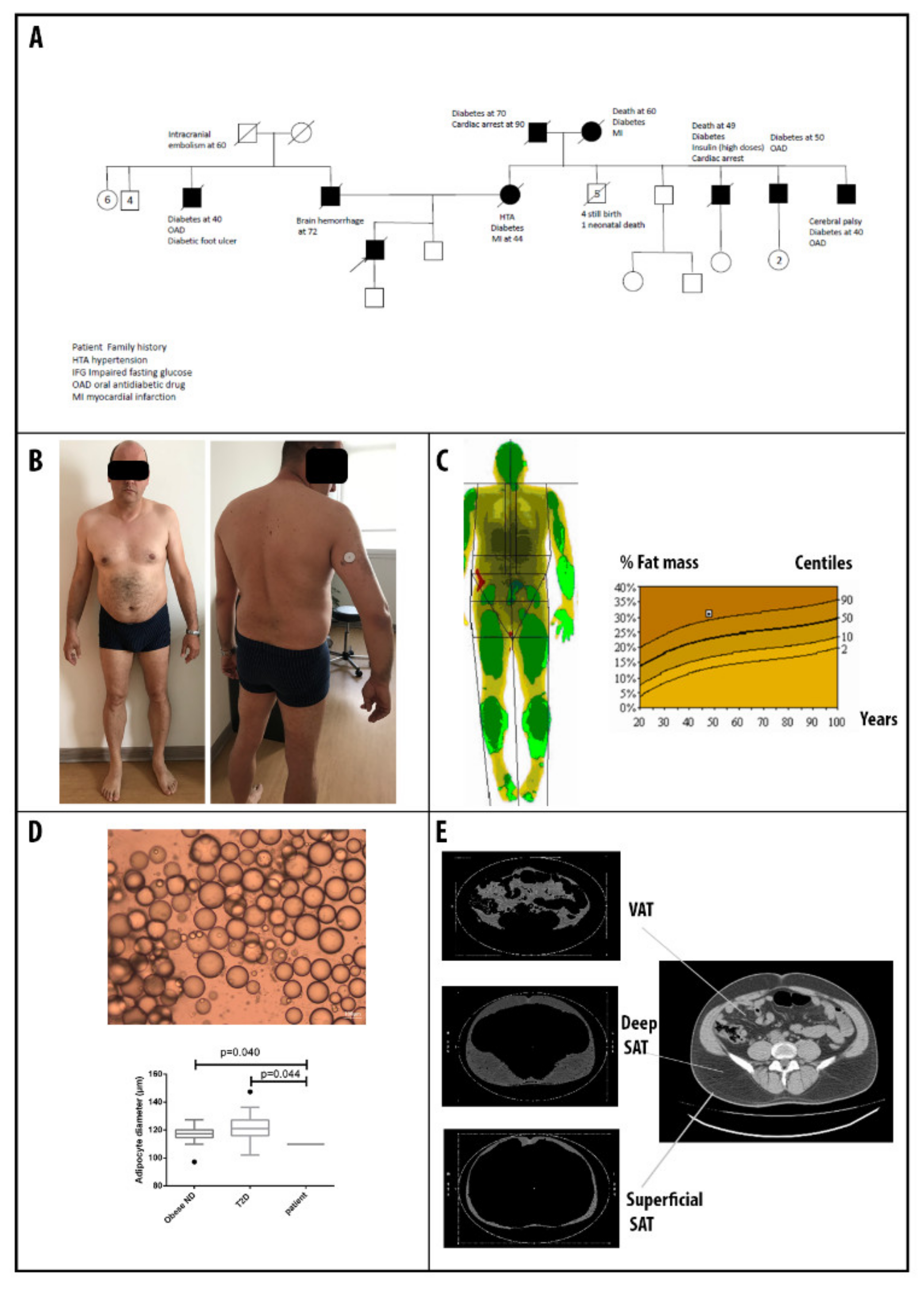
3.1. Patients’ Clinical Description
3.2. Molecular Findings
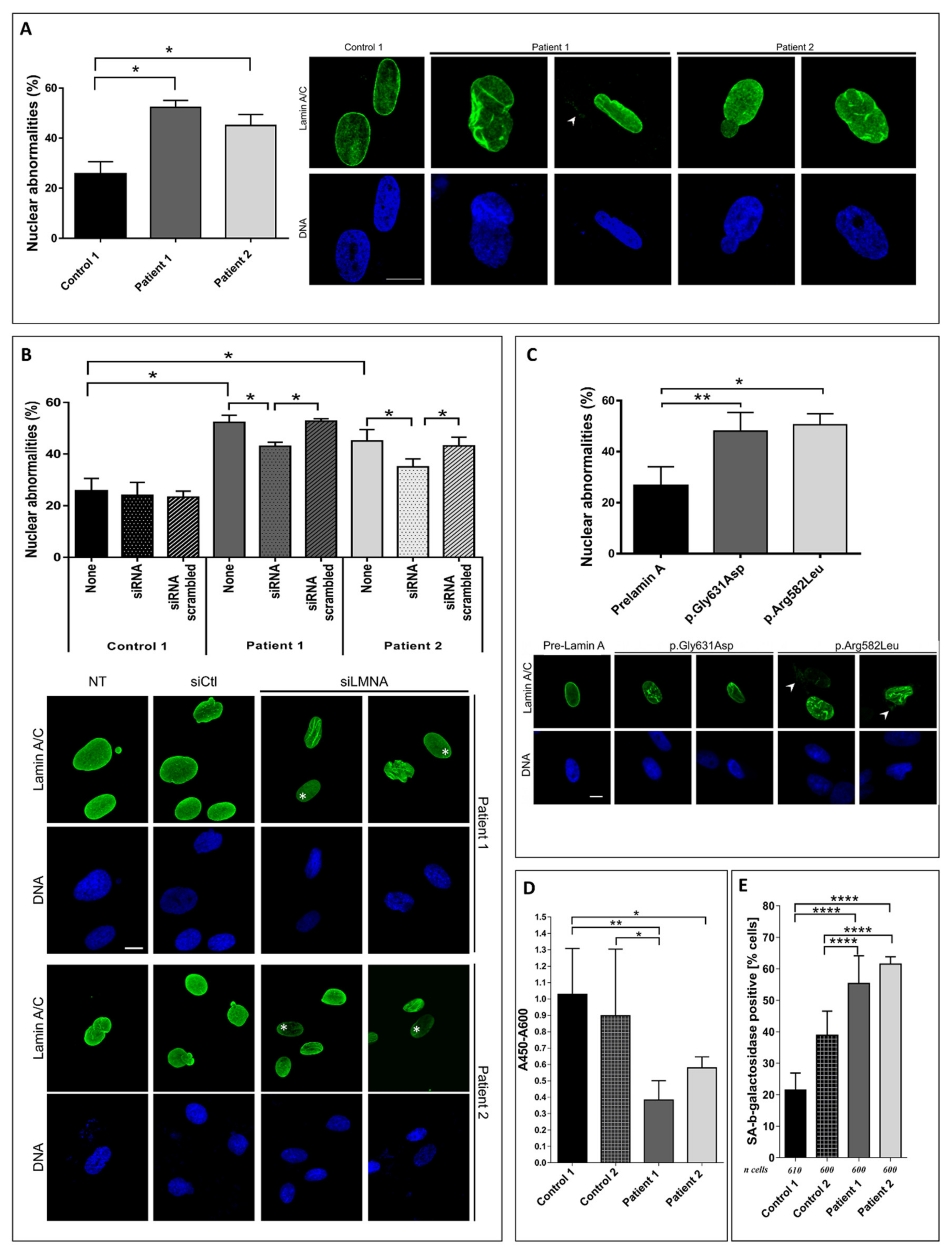
3.3. Patient Fibroblasts Functional Analysis
3.4. RNA Sequencing Analysis in Adipose Tissue
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vatier, C.; Vantyghem, M.-C.; Storey, C.; Jéru, I.; Christin-Maitre, S.; Fève, B.; Lascols, O.; Beltrand, J.; Carel, J.-C.; Vigouroux, C.; et al. Monogenic forms of lipodystrophic syndromes: Diagnosis, detection, and practical management considerations from clinical cases. Curr. Med. Res. Opin. 2019, 35, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Patni, N.; Garg, A. Lipodystrophies, dyslipidaemias and atherosclerotic cardiovascular disease. Pathology 2019, 51, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.J.; Araujo-Vilar, D.; Cheung, P.T.; Dunger, D.; Garg, A.; Jack, M.; Mungai, L.; Oral, E.A.; Patni, N.; Rother, K.I.; et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 4500–4511. [Google Scholar] [CrossRef] [PubMed]
- Vatier, C.; Bidault, G.; Briand, N.; Guénantin, A.-C.; Teyssières, L.; Lascols, O.; Capeau, J.; Vigouroux, C. What the genetics of lipodystrophy can teach us about insulin resistance and diabetes. Curr. Diab. Rep. 2013, 13, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Mounkes, L.; Kozlov, S.; Burke, B.; Stewart, C.L. The laminopathies: Nuclear structure meets disease. Curr. Opin. Genet. Dev. 2003, 13, 223–230. [Google Scholar] [CrossRef]
- Vigouroux, C.; Auclair, M.; Dubosclard, E.; Pouchelet, M.; Capeau, J.; Courvalin, J.C.; Buendia, B. Nuclear envelope disorganization in fibroblasts from lipodystrophic patients with heterozygous R482Q/W mutations in the lamin A/C gene. J. Cell. Sci. 2001, 114, 4459–4468. [Google Scholar] [PubMed]
- Muchir, A.; Medioni, J.; Laluc, M.; Massart, C.; Arimura, T.; van der Kooi, A.J.; Desguerre, I.; Mayer, M.; Ferrer, X.; Briault, S.; et al. Nuclear envelope alterations in fibroblasts from patients with muscular dystrophy, cardiomyopathy, and partial lipodystrophy carrying lamin A/C gene mutations. Muscle Nerve 2004, 30, 444–450. [Google Scholar] [CrossRef]
- Gruenbaum, Y.; Foisner, R. Lamins: Nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu. Rev. Biochem. 2015, 84, 131–164. [Google Scholar] [CrossRef]
- Guillín-Amarelle, C.; Fernández-Pombo, A.; Sánchez-Iglesias, S.; Araújo-Vilar, D. Lipodystrophic laminopathies: Diagnostic clues. Nucleus 2018, 9, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Decaudain, A.; Vantyghem, M.-C.; Guerci, B.; Hécart, A.-C.; Auclair, M.; Reznik, Y.; Narbonne, H.; Ducluzeau, P.-H.; Donadille, B.; Lebbé, C.; et al. New metabolic phenotypes in laminopathies: LMNA mutations in patients with severe metabolic syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 4835–4844. [Google Scholar] [CrossRef] [Green Version]
- Gonzaga-Jauregui, C.; Ge, W.; Staples, J.; Van Hout, C.; Yadav, A.; Colonie, R.; Leader, J.B.; Kirchner, H.L.; Geisinger-Regeneron DiscovEHR Collaboration, Regeneron Genetics Center; Murray, M.F.; et al. Clinical and Molecular Prevalence of Lipodystrophy in an Unascertained Large Clinical Care Cohort. Diabetes 2019. [Google Scholar]
- Vantyghem, M.C.; Vincent-Desplanques, D.; Defrance-Faivre, F.; Capeau, J.; Fermon, C.; Valat, A.S.; Lascols, O.; Hecart, A.C.; Pigny, P.; Delemer, B.; et al. Fertility and obstetrical complications in women with LMNA-related familial partial lipodystrophy. J. Clin. Endocrinol. Metab. 2008, 93, 2223–2229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galant, D.; Gaborit, B.; Desgrouas, C.; Abdesselam, I.; Bernard, M.; Levy, N.; Merono, F.; Coirault, C.; Roll, P.; Lagarde, A.; et al. A Heterozygous ZMPSTE24 Mutation Associated with Severe Metabolic Syndrome, Ectopic Fat Accumulation, and Dilated Cardiomyopathy. Cells 2016, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Dutour, A.; Roll, P.; Gaborit, B.; Courrier, S.; Alessi, M.-C.; Tregouet, D.-A.; Angelis, F.; Robaglia-Schlupp, A.; Lesavre, N.; Cau, P.; et al. High prevalence of laminopathies among patients with metabolic syndrome. Hum. Mol. Genet. 2011, 20, 3779–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonello-Palot, N.; Simoncini, S.; Robert, S.; Bourgeois, P.; Sabatier, F.; Levy, N.; Dignat-George, F.; Badens, C. Prelamin A accumulation in endothelial cells induces premature senescence and functional impairment. Atherosclerosis 2014, 237, 45–52. [Google Scholar] [CrossRef]
- Grundy, S.M.; Cleeman, J.I.; Daniels, S.R.; Donato, K.A.; Eckel, R.H.; Franklin, B.A.; Gordon, D.J.; Krauss, R.M.; Savage, P.J.; Smith, S.C.; et al. Diagnosis and management of the metabolic syndrome: An American Heart Association/National Heart, Lung, and Blood Institute scientific statement: Executive Summary. Crit. Pathw. Cardiol. 2005, 4, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Salgado, D.; Desvignes, J.-P.; Rai, G.; Blanchard, A.; Miltgen, M.; Pinard, A.; Lévy, N.; Collod-Béroud, G.; Béroud, C. UMD-Predictor: A High-Throughput Sequencing Compliant System for Pathogenicity Prediction of any Human cDNA Substitution. Hum. Mutat. 2016, 37, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Pongsakul, N.; Vinaiphat, A.; Chanchaem, P.; Fong-Ngern, K.; Thongboonkerd, V. Lamin A/C in renal tubular cells is important for tissue repair, cell proliferation, and calcium oxalate crystal adhesion, and is associated with potential crystal receptors. FASEB J. 2016, 30, 3368–3377. [Google Scholar] [CrossRef] [Green Version]
- Caron, M.; Auclair, M.; Donadille, B.; Béréziat, V.; Guerci, B.; Laville, M.; Narbonne, H.; Bodemer, C.; Lascols, O.; Capeau, J.; et al. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ. 2007, 14, 1759–1767. [Google Scholar] [CrossRef]
- Ambonville, C.; Bouldouyre, M.-A.; Laforêt, P.; Richard, P.; Benveniste, O.; Vigouroux, C. A complex case of diabetes due to LMNA mutation. Rev. Med. Interne 2017, 38, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Florwick, A.; Dharmaraj, T.; Jurgens, J.; Valle, D.; Wilson, K.L. LMNA Sequences of 60,706 Unrelated Individuals Reveal 132 Novel Missense Variants in A-Type Lamins and Suggest a Link between Variant p.G602S and Type 2 Diabetes. Front. Genet. 2017, 8, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeru, I.; Vatier, C.; Vantyghem, M.-C.; Lascols, O.; Vigouroux, C. LMNA-associated partial lipodystrophy: Anticipation of metabolic complications. J. Med. Genet. 2017, 54, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Bayona, P.W.; Kim, M.; Chang, J.; Hong, S.; Park, Y.; Budiman, A.; Kim, Y.-J.; Choi, C.Y.; Kim, W.S.; et al. Macrophage Lamin A/C Regulates Inflammation and the Development of Obesity-Induced Insulin Resistance. Front. Immunol 2018, 9, 696. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, J.; Wei, Y.; Zhang, W.; Geng, M.; Yuan, Y.; Chen, Y.; Sun, Y.; Chen, H.; Zhang, Y.; et al. Fat-Specific Knockout of Mecp2 Upregulates Slpi to Reduce Obesity by Enhancing Browning. Diabetes 2019. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, F.; Evangelisti, C.; Cenni, V.; Fazio, A.; Paganelli, F.; Martelli, A.M.; Lattanzi, G. The Cutting Edge: The Role of mTOR Signaling in Laminopathies. Int J. Mol. Sci. 2019, 20, 847. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desgrouas, C.; Varlet, A.-A.; Dutour, A.; Galant, D.; Merono, F.; Bonello-Palot, N.; Bourgeois, P.; Lasbleiz, A.; Petitjean, C.; Ancel, P.; et al. Unraveling LMNA Mutations in Metabolic Syndrome: Cellular Phenotype and Clinical Pitfalls. Cells 2020, 9, 310. https://doi.org/10.3390/cells9020310
Desgrouas C, Varlet A-A, Dutour A, Galant D, Merono F, Bonello-Palot N, Bourgeois P, Lasbleiz A, Petitjean C, Ancel P, et al. Unraveling LMNA Mutations in Metabolic Syndrome: Cellular Phenotype and Clinical Pitfalls. Cells. 2020; 9(2):310. https://doi.org/10.3390/cells9020310
Chicago/Turabian StyleDesgrouas, Camille, Alice-Anaïs Varlet, Anne Dutour, Damien Galant, Françoise Merono, Nathalie Bonello-Palot, Patrice Bourgeois, Adèle Lasbleiz, Cathy Petitjean, Patricia Ancel, and et al. 2020. "Unraveling LMNA Mutations in Metabolic Syndrome: Cellular Phenotype and Clinical Pitfalls" Cells 9, no. 2: 310. https://doi.org/10.3390/cells9020310