Prolonged Hyperoxygenation Treatment Improves Vein Graft Patency and Decreases Macrophage Content in Atherosclerotic Lesions in ApoE3*Leiden Mice

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Vein Graft Surgery
2.3. Carbogen Treatment
2.4. Histological and Immunohistochemical Assessment of Vein Grafts
2.5. RNA Isolation, cDNA Synthesis and qPCR
2.6. Bone Marrow Derived Macrophages Isolation and In Vitro Experiments
2.7. Statistical Analysis
3. Results
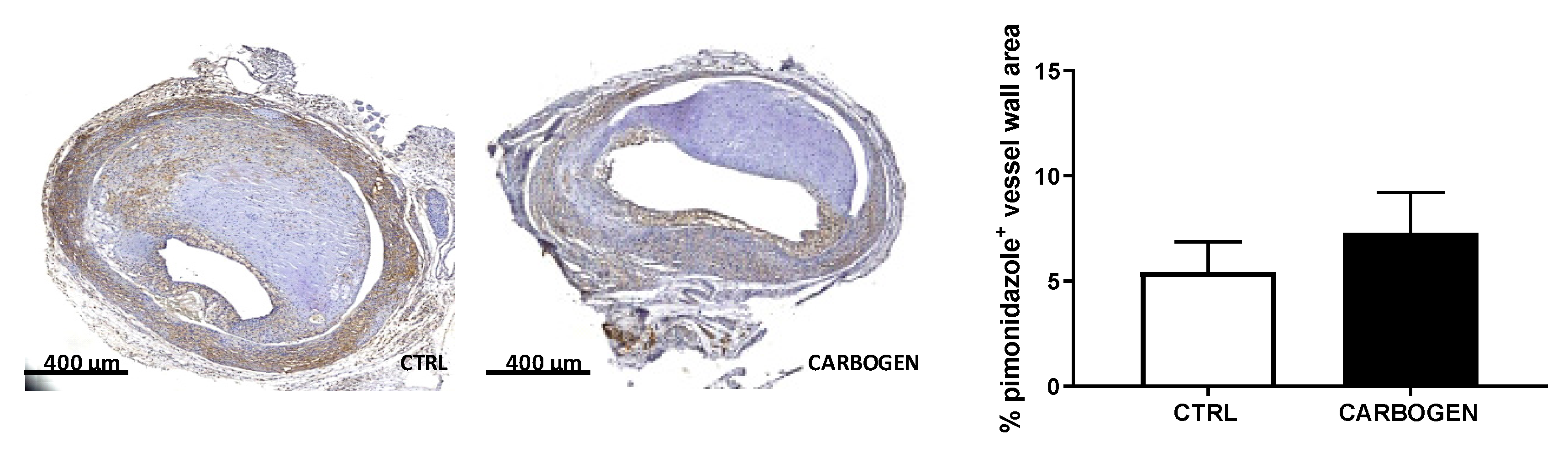
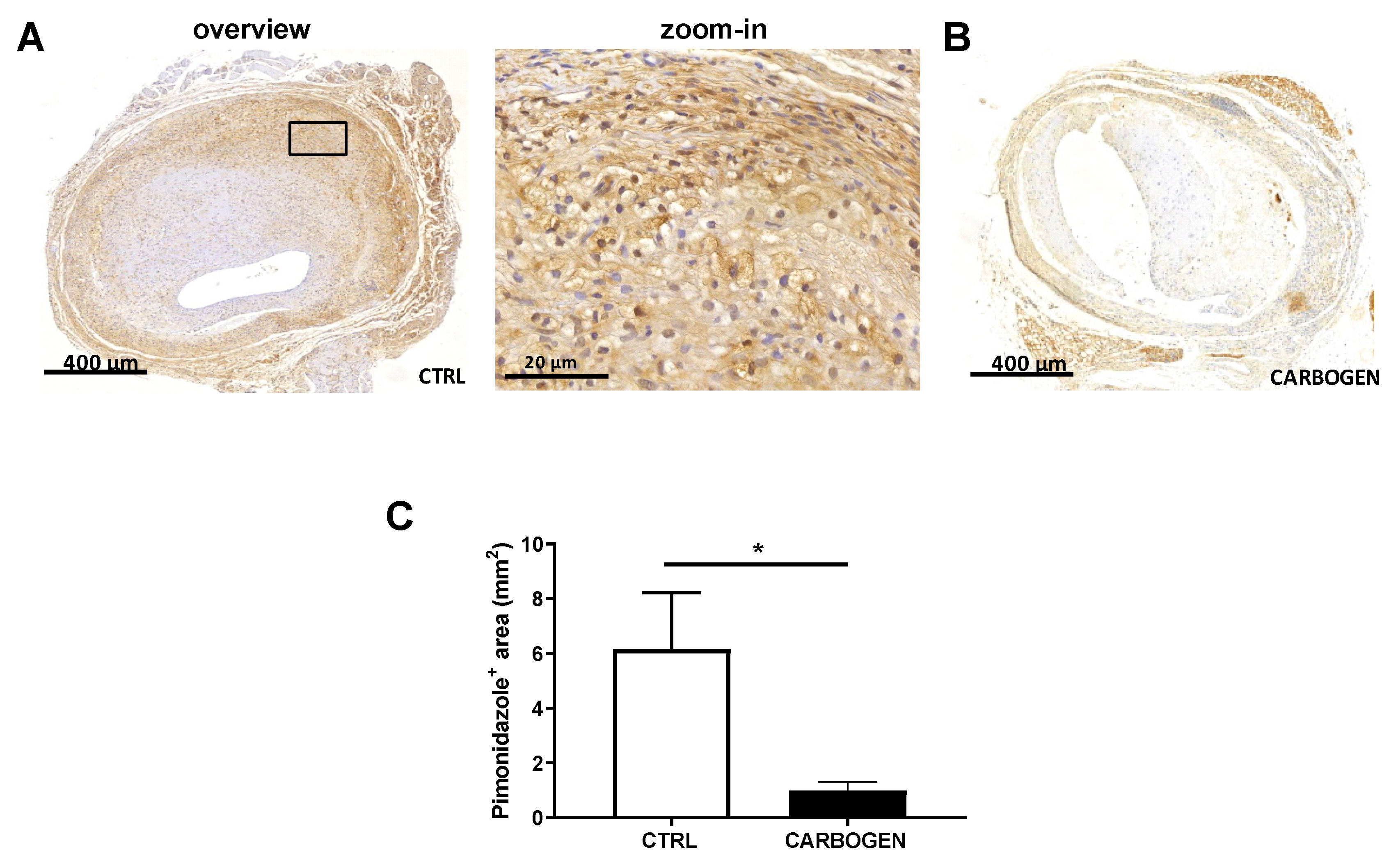
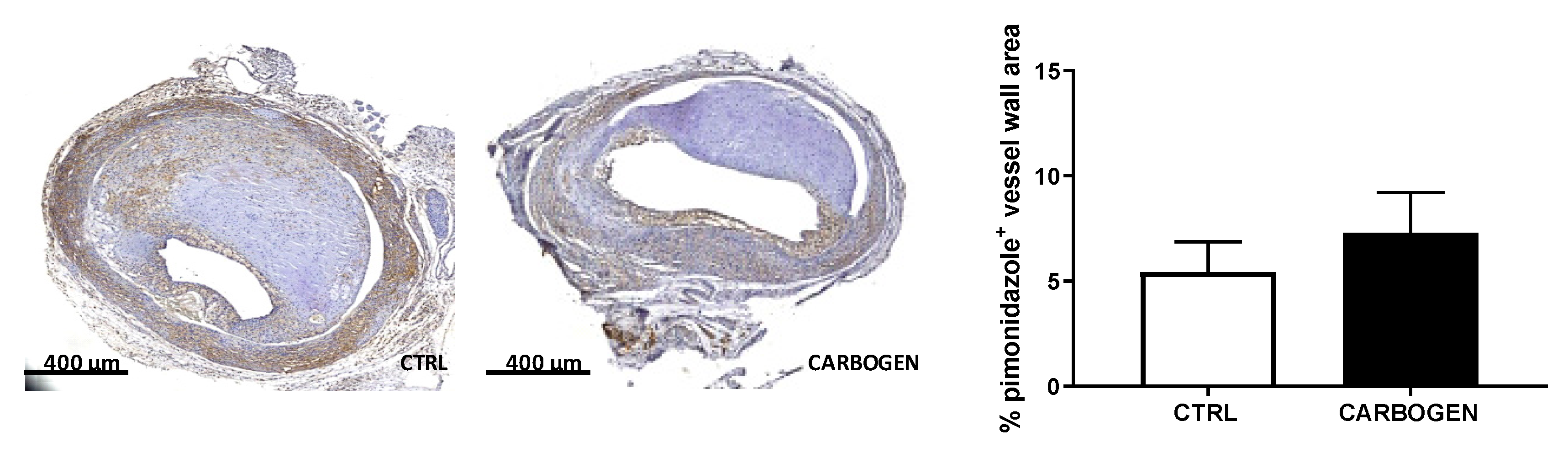
3.1. Acute Carbogen Exposure Reduces Intraplaque Hypoxia
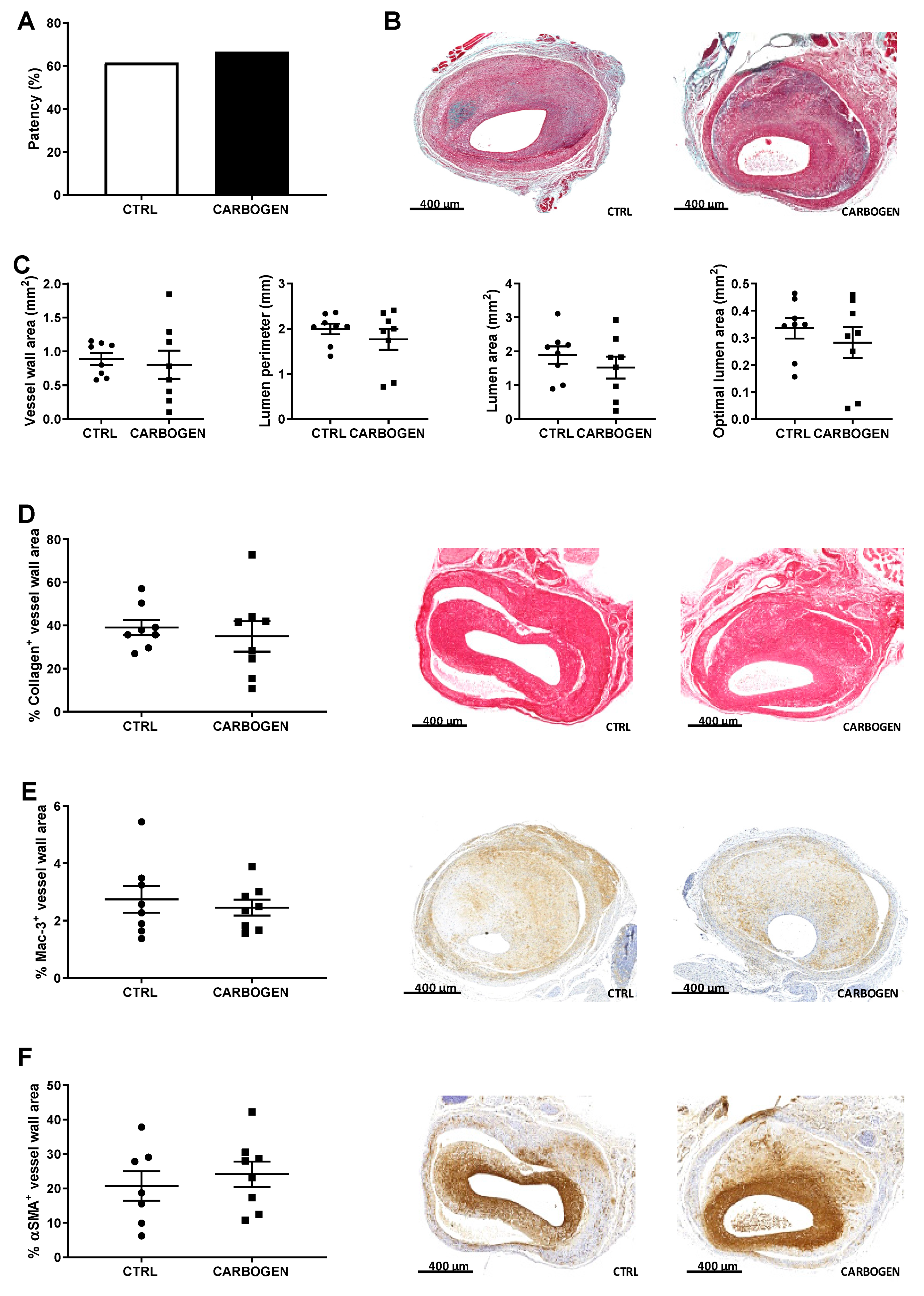
3.2. Chronic Carbogen Exposure Does not Influence Intraplaque Hypoxia
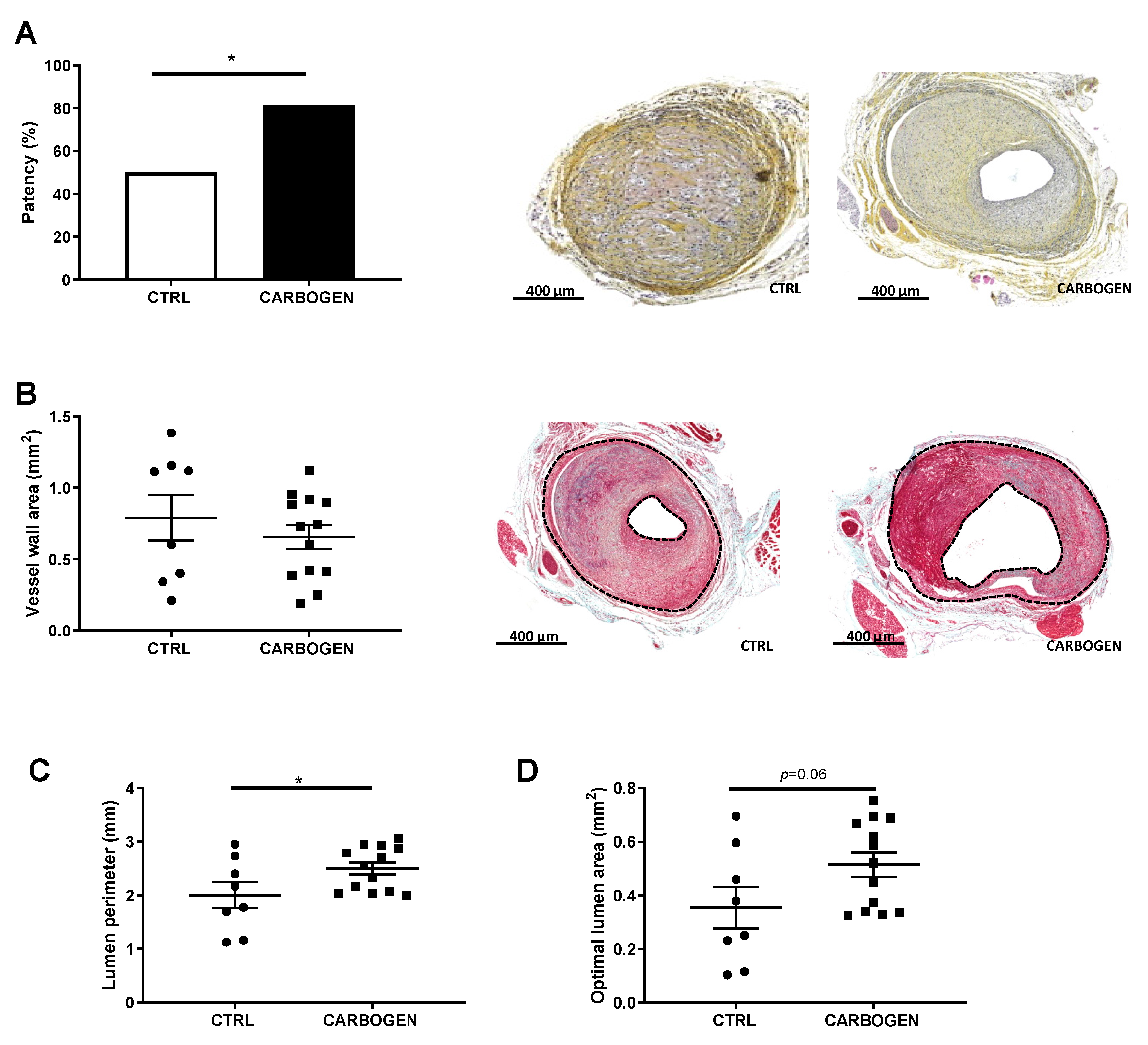
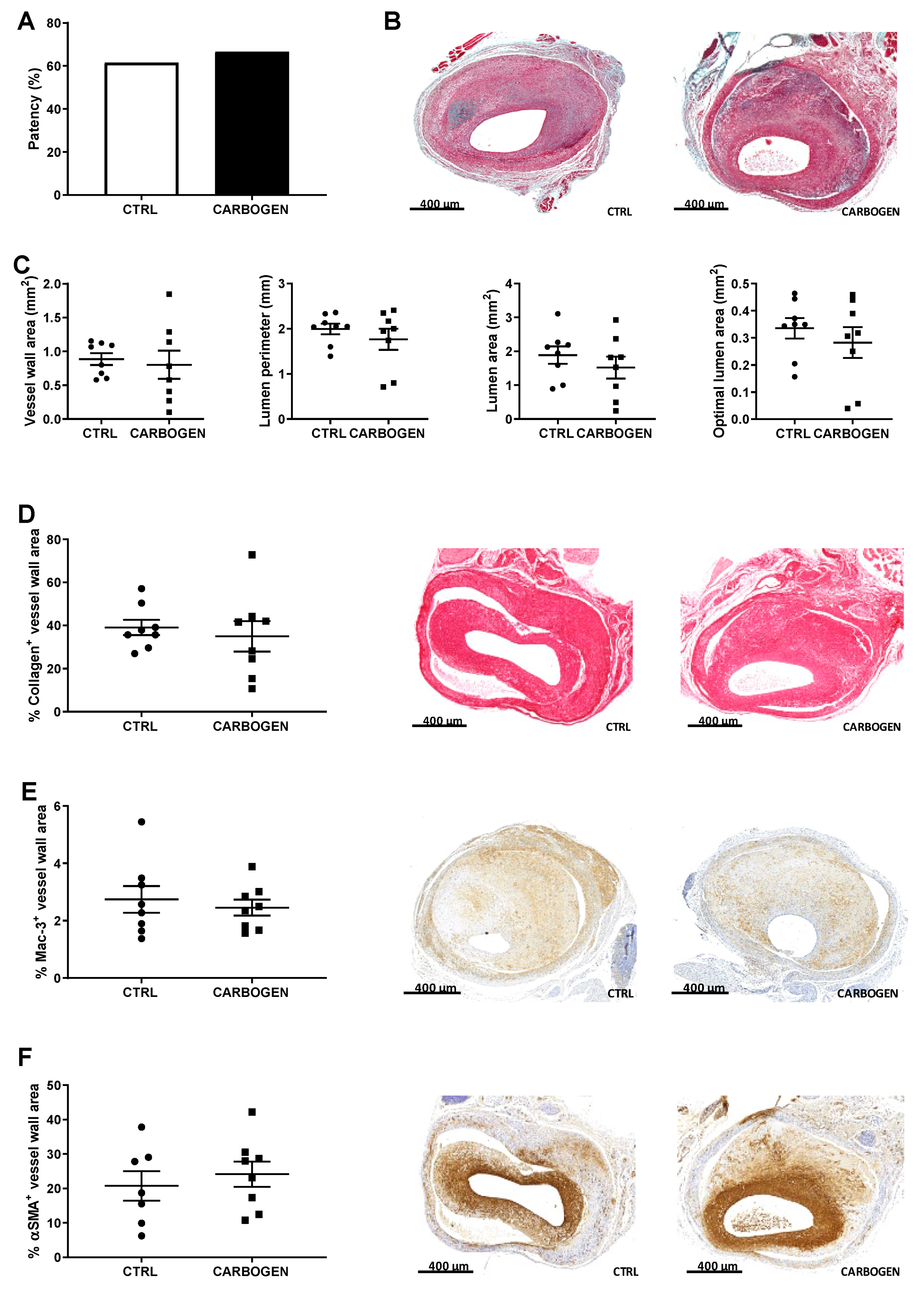
3.3. Chronic Exposure to Carbogen Plays a Protective Role Against Occlusions
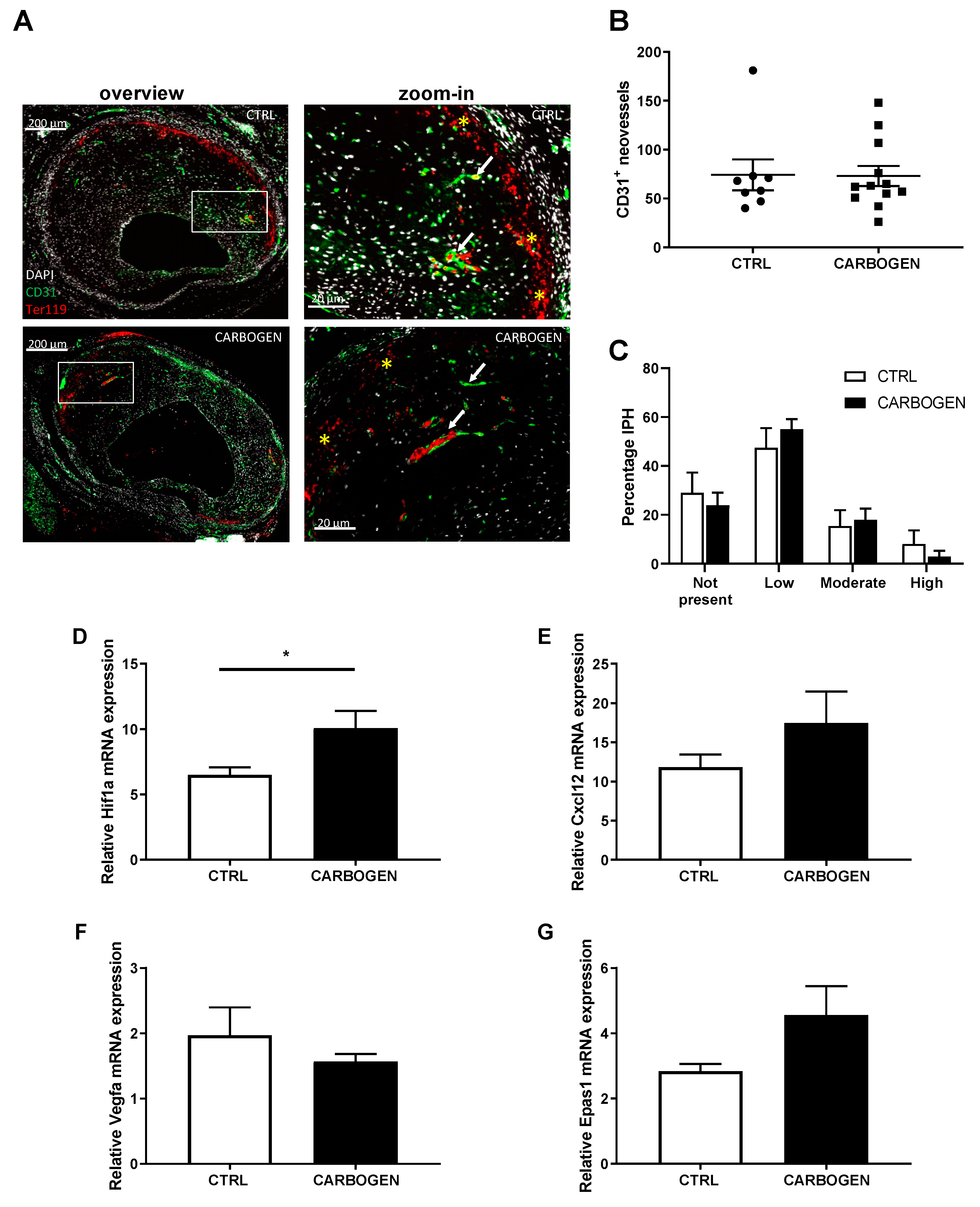
3.4. Chronic Carbogen Treatment Does Not Have an Effect on Intraplaque Angiogenesis and Intraplaque Hemorrhage
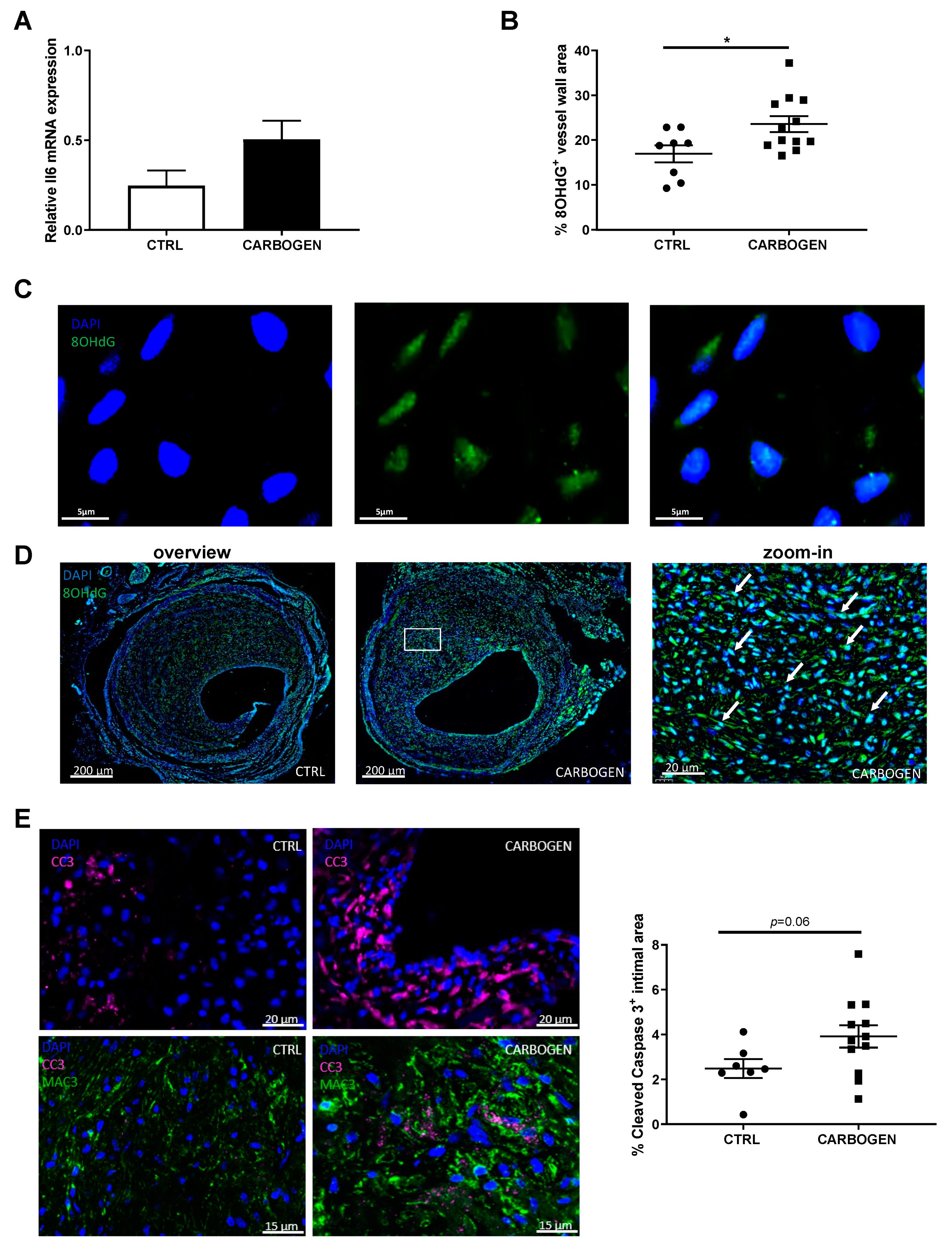
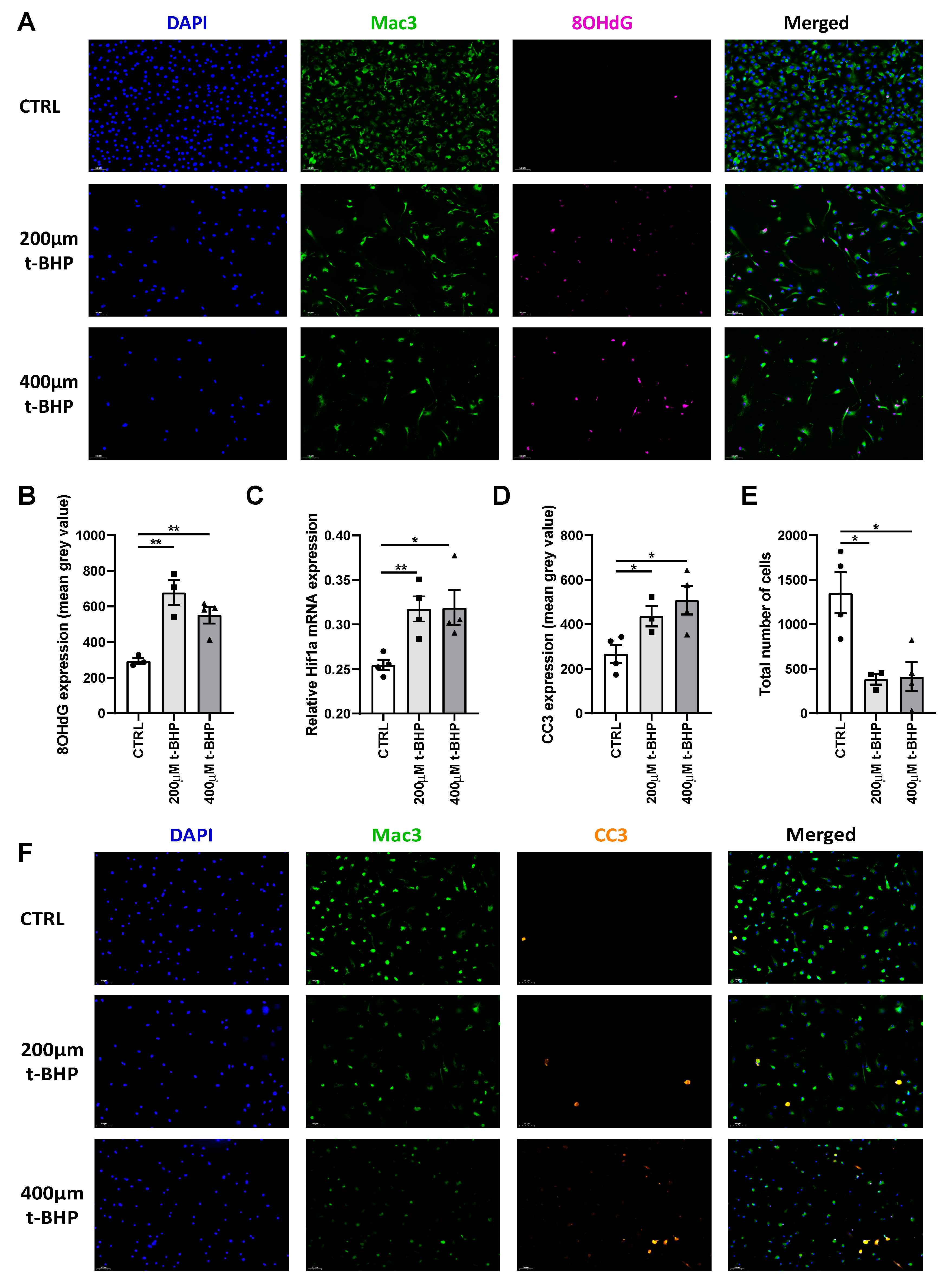
3.5. Chronic Carbogen Treatment Induces Accumulation of Reactive Oxygen Species and Apoptosis
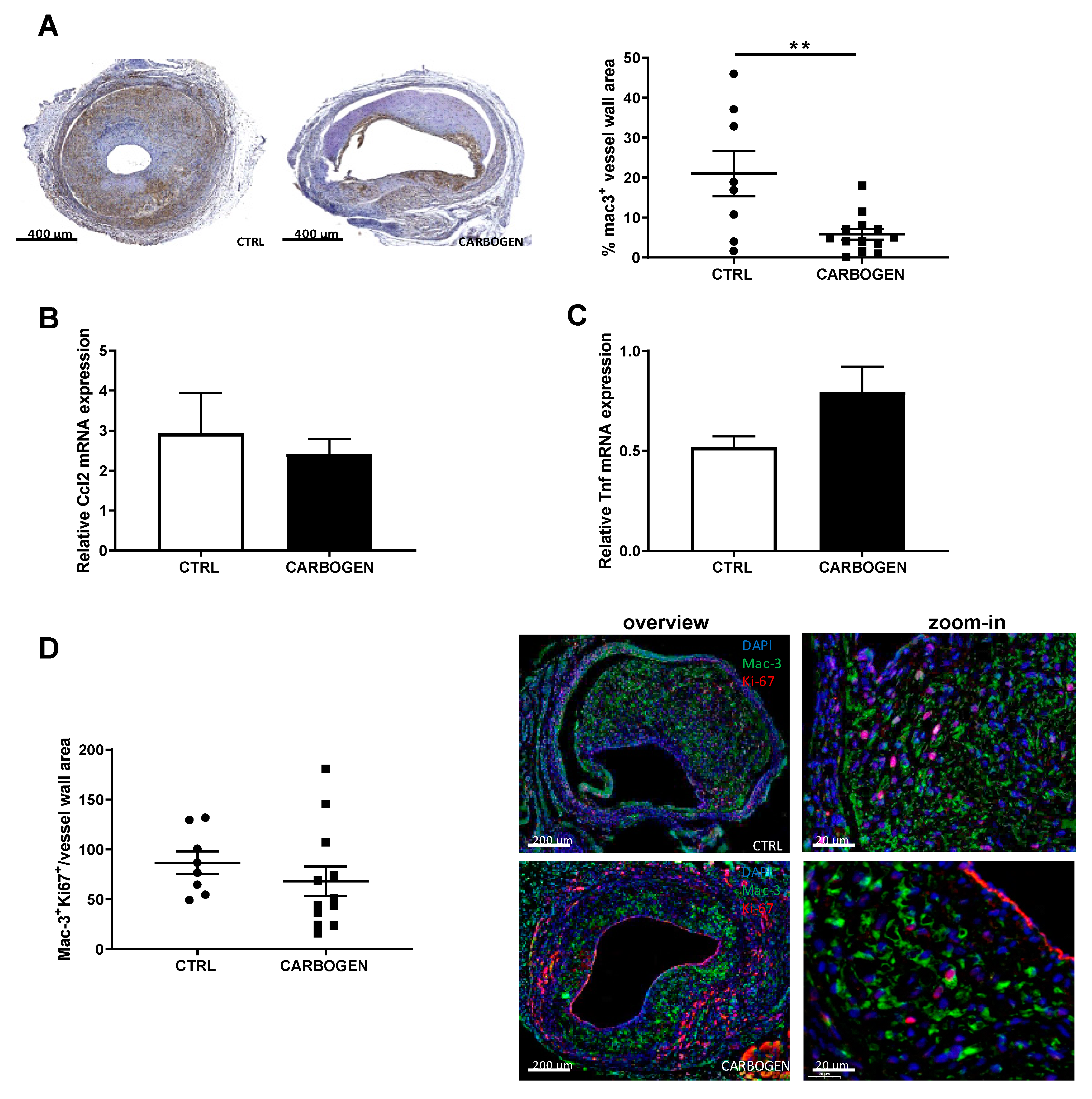
3.6. Chronic Carbogen Exposure Reduces Inflammatory Cell Content
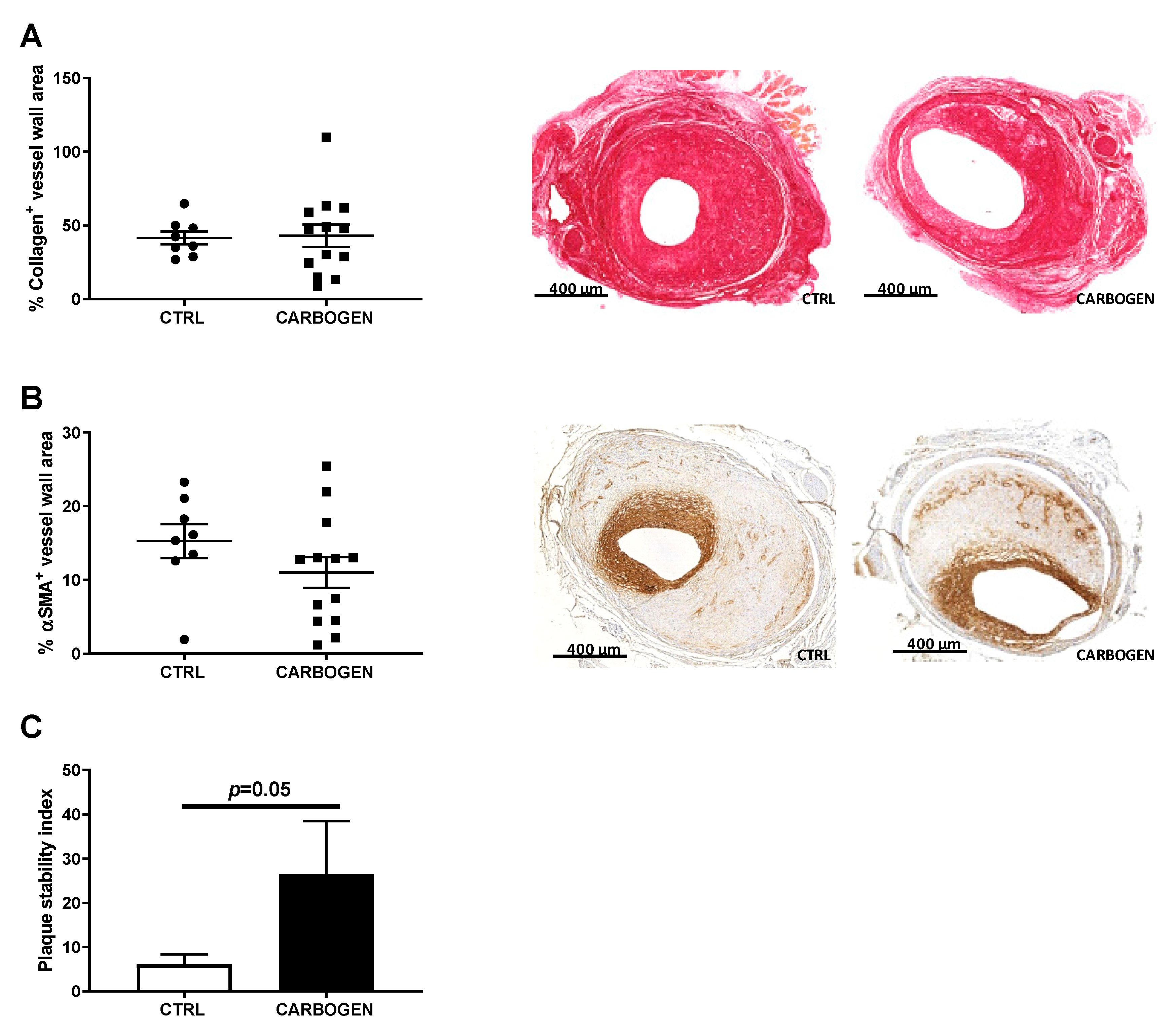
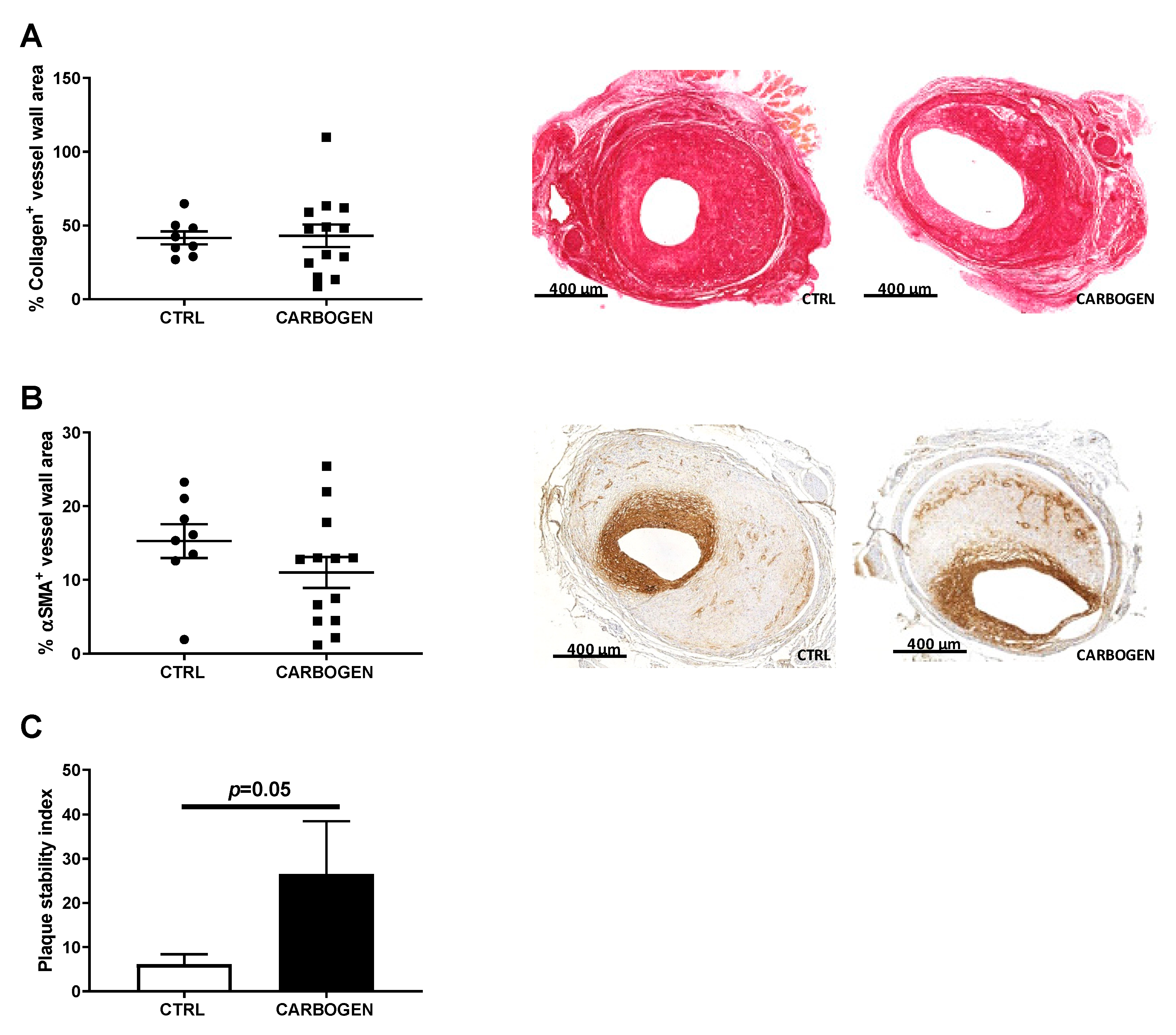
3.7. Chronic Carbogen Treatment Does Not Affect Plaque Size but Increases Plaque Stability
3.8. ROS Increases DNA Damage and Apoptosis in Bone Marrow Derived Macrophages In Vitro
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- de Vries, M.R.; Quax, P.H. Plaque angiogenesis and its relation to inflammation and atherosclerotic plaque destabilization. Curr. Opin. Lipidol. 2016, 27, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Bauer, G.E.; Caldwell, M.P.; Santilli, S.M. Association of artery wall hypoxia and cellular proliferation at a vascular anastomosis. J. Surg. Res. 2000, 91, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Sluimer, J.C.; Gasc, J.M.; van Wanroij, J.L.; Kisters, N.; Groeneweg, M.; Sollewijn Gelpke, M.D.; Cleutjens, J.P.; van den Akker, L.H.; Corvol, P.; Wouters, B.G.; et al. Hypoxia, hypoxia-inducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. J. Am. Coll. Cardiol. 2008, 51, 1258–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, M.R.; Parma, L.; Peters, H.A.B.; Schepers, A.; Hamming, J.F.; Jukema, J.W.; Goumans, M.; Guo, L.; Finn, A.V.; Virmani, R.; et al. Blockade of vascular endothelial growth factor receptor 2 inhibits intraplaque haemorrhage by normalization of plaque neovessels. J. Intern. Med. 2019, 285, 59–74. [Google Scholar] [CrossRef]
- Marsch, E.; Sluimer, J.C.; Daemen, M.J. Hypoxia in atherosclerosis and inflammation. Curr. Opin. Lipidol. 2013, 24, 393–400. [Google Scholar] [CrossRef]
- Subbotin, V.M. Excessive intimal hyperplasia in human coronary arteries before intimal lipid depositions is the initiation of coronary atherosclerosis and constitutes a therapeutic target. Drug Discov. Today 2016, 21, 1578–1595. [Google Scholar] [CrossRef] [Green Version]
- Jain, T.; Nikolopoulou, E.A.; Xu, Q.; Qu, A. Hypoxia inducible factor as a therapeutic target for atherosclerosis. Pharmacol. Ther. 2018, 183, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Balogh, E.; Toth, A.; Mehes, G.; Trencsenyi, G.; Paragh, G.; Jeney, V. Hypoxia Triggers Osteochondrogenic Differentiation of Vascular Smooth Muscle Cells in an HIF-1 (Hypoxia-Inducible Factor 1)-Dependent and Reactive Oxygen Species-Dependent Manner. Arter. Thromb. Vasc. Biol. 2019, 39, 1088–1099. [Google Scholar] [CrossRef]
- Parma, L.; Baganha, F.; Quax, P.H.A.; de Vries, M.R. Plaque angiogenesis and intraplaque hemorrhage in atherosclerosis. Eur. J. Pharm. 2017, 816, 107–115. [Google Scholar] [CrossRef]
- Sluimer, J.C.; Kolodgie, F.D.; Bijnens, A.P.; Maxfield, K.; Pacheco, E.; Kutys, B.; Duimel, H.; Frederik, P.M.; van Hinsbergh, V.W.; Virmani, R.; et al. Thin-walled microvessels in human coronary atherosclerotic plaques show incomplete endothelial junctions relevance of compromised structural integrity for intraplaque microvascular leakage. J. Am. Coll. Cardiol. 2009, 53, 1517–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aarup, A.; Pedersen, T.X.; Junker, N.; Christoffersen, C.; Bartels, E.D.; Madsen, M.; Nielsen, C.H.; Nielsen, L.B. Hypoxia-Inducible Factor-1alpha Expression in Macrophages Promotes Development of Atherosclerosis. Arter. Thromb. Vasc. Biol. 2016, 36, 1782–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hultén, L.M.; Levin, M. The role of hypoxia in atherosclerosis. Curr. Opin. Lipidol. 2009, 20, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Semba, H.; Takeda, N. The Roles of Hypoxia Signaling in the Pathogenesis of Cardiovascular Diseases. J. Atheroscler. Thromb. 2017, 24, 884–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsch, E.; Theelen, T.L.; Demandt, J.A.; Jeurissen, M.; van Gink, M.; Verjans, R.; Janssen, A.; Cleutjens, J.P.; Meex, S.J.; Donners, M.M.; et al. Reversal of hypoxia in murine atherosclerosis prevents necrotic core expansion by enhancing efferocytosis. Arter. Thromb. Vasc. Biol. 2014, 34, 2545–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, M.R.; Niessen, H.W.; Lowik, C.W.; Hamming, J.F.; Jukema, J.W.; Quax, P.H. Plaque rupture complications in murine atherosclerotic vein grafts can be prevented by TIMP-1 overexpression. PLoS ONE 2012, 7, e47134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamieson, D.; Chance, B.; Cadenas, E.; Boveris, A. The relation of free radical production to hyperoxia. Annu. Rev. Physiol. 1986, 48, 703–719. [Google Scholar] [CrossRef]
- Simons, K.H.; de Vries, M.R.; Peters, H.A.B.; Hamming, J.F.; Jukema, J.W.; Quax, P.H.A. The protective role of Toll-like receptor 3 and type-I interferons in the pathophysiology of vein graft disease. J. Mol. Cell. Cardiol. 2018, 121, 16–24. [Google Scholar] [CrossRef]
- Han, L.; Wang, Y.L.; Sun, Y.C.; Hu, Z.Y.; Hu, K.; Du, L.B. tert-Butylhydroperoxide induces apoptosis in RAW264.7 macrophages via a mitochondria-mediated signaling pathway. Toxicol. Res. 2018, 7, 970–976. [Google Scholar] [CrossRef] [Green Version]
- Yip, K.; Alonzi, R. Carbogen gas and radiotherapy outcomes in prostate cancer. Ther. Adv. Urol. 2013, 5, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.J.; Zhang, Z.; Xu, J.; Jin, N.; Luo, S.; Larson, A.C.; Lu, G.M. Carbogen gas-challenge blood oxygen level-dependent magnetic resonance imaging in hepatocellular carcinoma: Initial results. Oncol. Lett. 2015, 10, 2009–2014. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.G.; Khan, N.; Du, G.X.; Hodge, S.; Swartz, H.M. Temporal variation in the response of tumors to hyperoxia with breathing carbogen and oxygen. Med. Gas. Res. 2016, 6, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Chakhoyan, A.; Corroyer-Dulmont, A.; Leblond, M.M.; Gerault, A.; Toutain, J.; Chazaviel, L.; Divoux, D.; Petit, E.; MacKenzie, E.T.; Kauffmann, F.; et al. Carbogen-induced increases in tumor oxygenation depend on the vascular status of the tumor: A multiparametric MRI study in two rat glioblastoma models. J. Cereb. Blood Flow Metab. 2017, 37, 2270–2282. [Google Scholar] [CrossRef] [Green Version]
- de Vries, M.R.; Simons, K.H.; Jukema, J.W.; Braun, J.; Quax, P.H. Vein graft failure: From pathophysiology to clinical outcomes. Nat. Rev. Cardiol. 2016, 13, 451–470. [Google Scholar] [CrossRef] [PubMed]
- Fowler, S.B. Carbogen in the management of a central retinal artery occlusion. Insight (Am. Soc. Ophthalmic Regist. Nurses) 2012, 37, 10–11. [Google Scholar]
- Gore, A.; Muralidhar, M.; Espey, M.G.; Degenhardt, K.; Mantell, L.L. Hyperoxia sensing: From molecular mechanisms to significance in disease. J. Immunotoxicol. 2010, 7, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Tang, S.; Xin, J.; Wei, Y.; Liu, D. Reactive oxygen species induce injury of the intestinal epithelium during hyperoxia. Int. J. Mol. Med. 2018, 41, 322–330. [Google Scholar] [CrossRef] [Green Version]
- Petrache, I.; Choi, M.E.; Otterbein, L.E.; Chin, B.Y.; Mantell, L.L.; Horowitz, S.; Choi, A.M. Mitogen-activated protein kinase pathway mediates hyperoxia-induced apoptosis in cultured macrophage cells. Am. J. Physiol. 1999, 277, L589–L595. [Google Scholar] [CrossRef]
- Halliwell, B.; Chirico, S. Lipid peroxidation: Its mechanism, measurement, and significance. Am. J. Clin. Nutr. 1993, 57, 715S–724S, discussion 724S-725S. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.D.; Dizdaroglu, M.; Cooke, M.S. Oxidative DNA damage and disease: Induction, repair and significance. Mutat. Res. 2004, 567, 1–61. [Google Scholar] [CrossRef]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 11427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traenckner, E.B.; Pahl, H.L.; Henkel, T.; Schmidt, K.N.; Wilk, S.; Baeuerle, P.A. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J. 1995, 14, 2876–2883. [Google Scholar] [CrossRef] [PubMed]
- Michiels, C.; Minet, E.; Mottet, D.; Raes, M. Regulation of gene expression by oxygen: NF-kappaB and HIF-1, two extremes. Free Radic. Biol. Med. 2002, 33, 1231–1242. [Google Scholar] [CrossRef]
- Bonello, S.; Zahringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Gorlach, A. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arter. Thromb. Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef] [Green Version]
- Tawakol, A.; Singh, P.; Mojena, M.; Pimentel-Santillana, M.; Emami, H.; MacNabb, M.; Rudd, J.H.; Narula, J.; Enriquez, J.A.; Traves, P.G.; et al. HIF-1alpha and PFKFB3 Mediate a Tight Relationship Between Proinflammatory Activation and Anerobic Metabolism in Atherosclerotic Macrophages. Arter. Thromb. Vasc. Biol. 2015, 35, 1463–1471. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parma, L.; Peters, H.A.B.; Baganha, F.; Sluimer, J.C.; de Vries, M.R.; Quax, P.H.A. Prolonged Hyperoxygenation Treatment Improves Vein Graft Patency and Decreases Macrophage Content in Atherosclerotic Lesions in ApoE3*Leiden Mice. Cells 2020, 9, 336. https://doi.org/10.3390/cells9020336
Parma L, Peters HAB, Baganha F, Sluimer JC, de Vries MR, Quax PHA. Prolonged Hyperoxygenation Treatment Improves Vein Graft Patency and Decreases Macrophage Content in Atherosclerotic Lesions in ApoE3*Leiden Mice. Cells. 2020; 9(2):336. https://doi.org/10.3390/cells9020336
Chicago/Turabian StyleParma, Laura, Hendrika A. B. Peters, Fabiana Baganha, Judith C. Sluimer, Margreet R. de Vries, and Paul H.A. Quax. 2020. "Prolonged Hyperoxygenation Treatment Improves Vein Graft Patency and Decreases Macrophage Content in Atherosclerotic Lesions in ApoE3*Leiden Mice" Cells 9, no. 2: 336. https://doi.org/10.3390/cells9020336