Development and Differentiation in Monobodies Based on the Fibronectin Type 3 Domain


Abstract
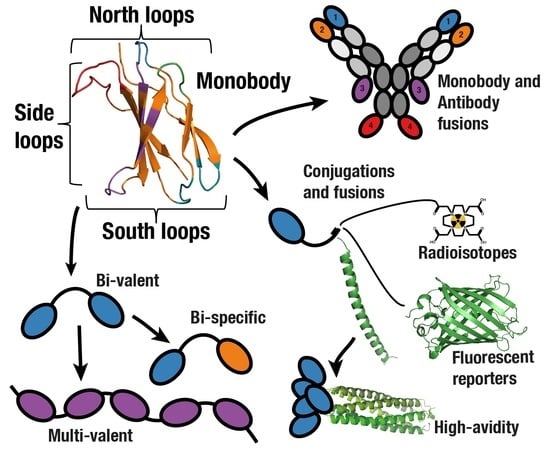
{kind=link}
{kind=link}
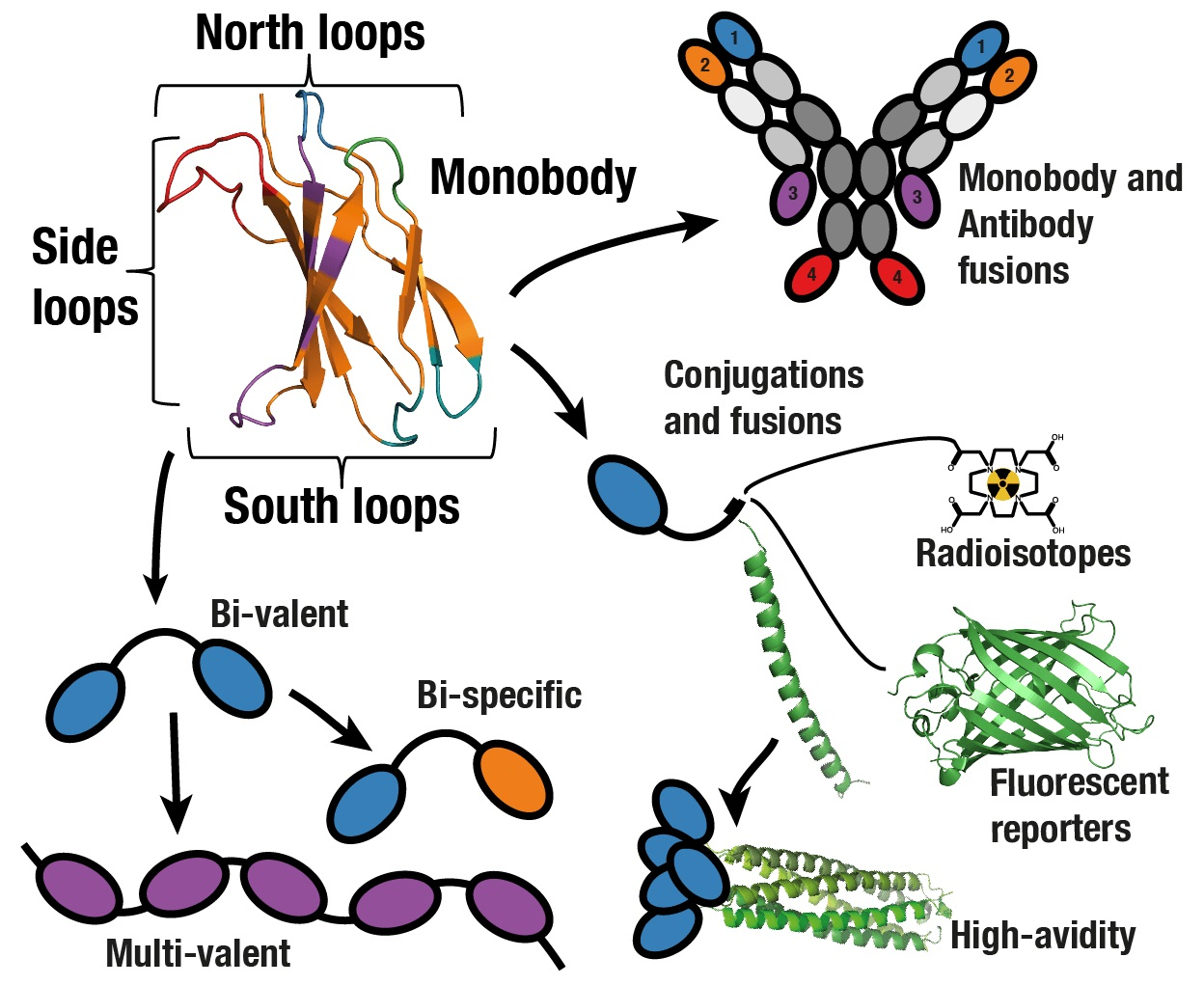
{kind=link}
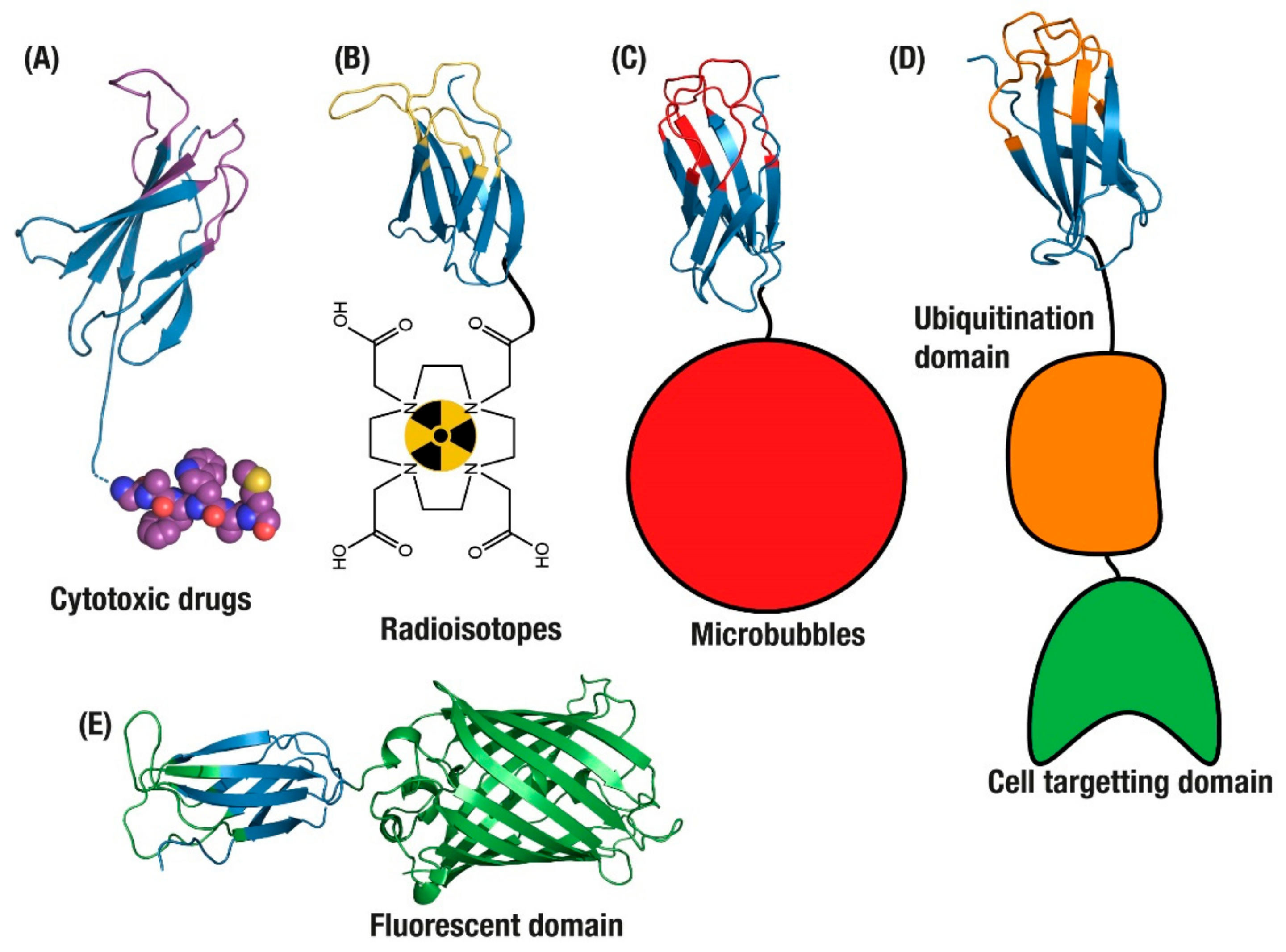
{kind=link}
{kind=link}
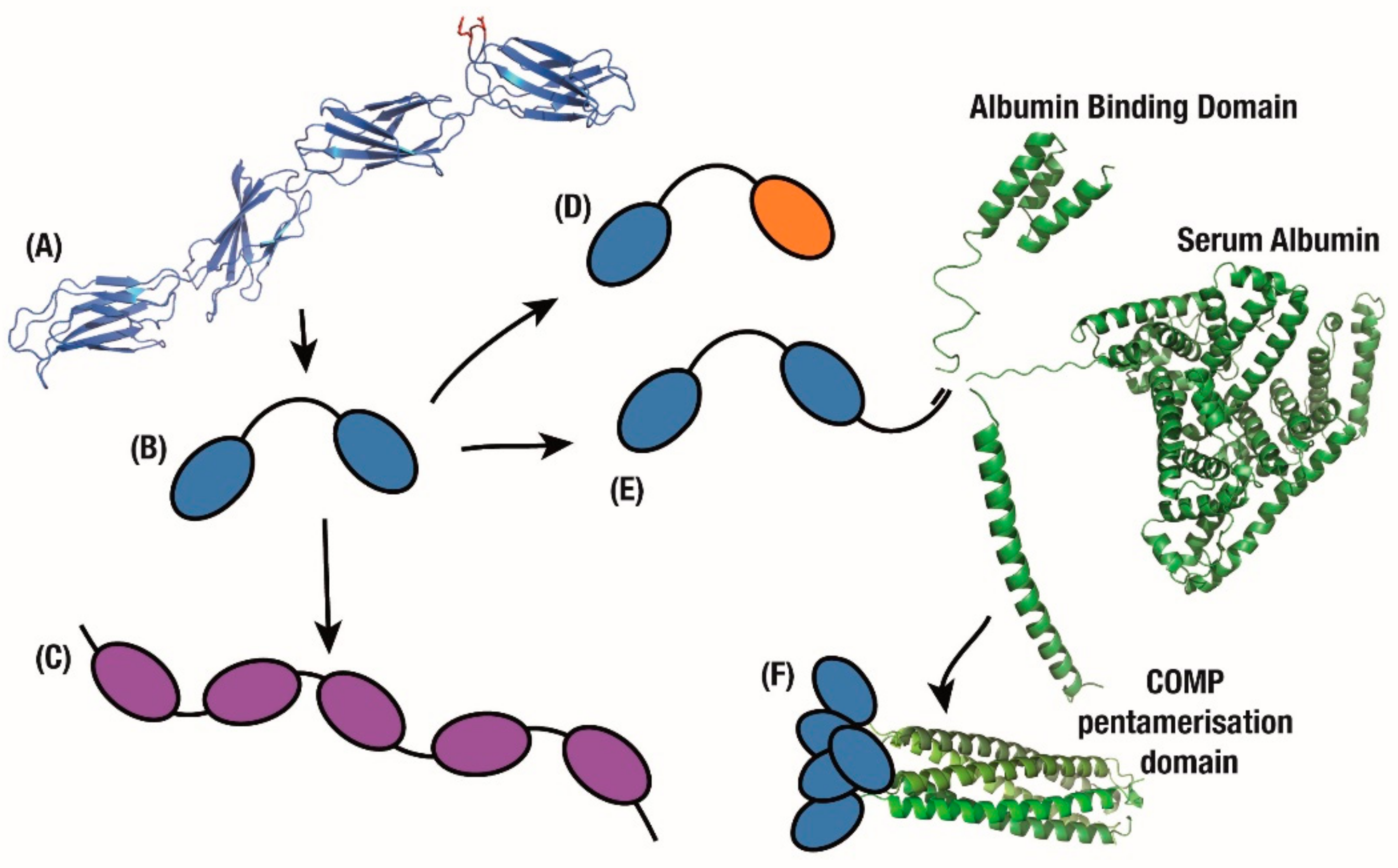
{kind=link}
1. Introduction
1.1. Monobodies Based on FN3
1.2. The Fibronectin Assembly Offers Functional Differentiation from Antibodies
1.3. Modern FN3 Derivatives in Development
1.4. Learning from the First Clinical Monobodies
2. Monomeric Monobodies in Therapeutics and Diagnostics
2.1. Delivery Agents
2.2. Biosensors
2.3. Intracellular Applications
3. Multidomain Monobodies
3.1. Fusion to Extend Half-life
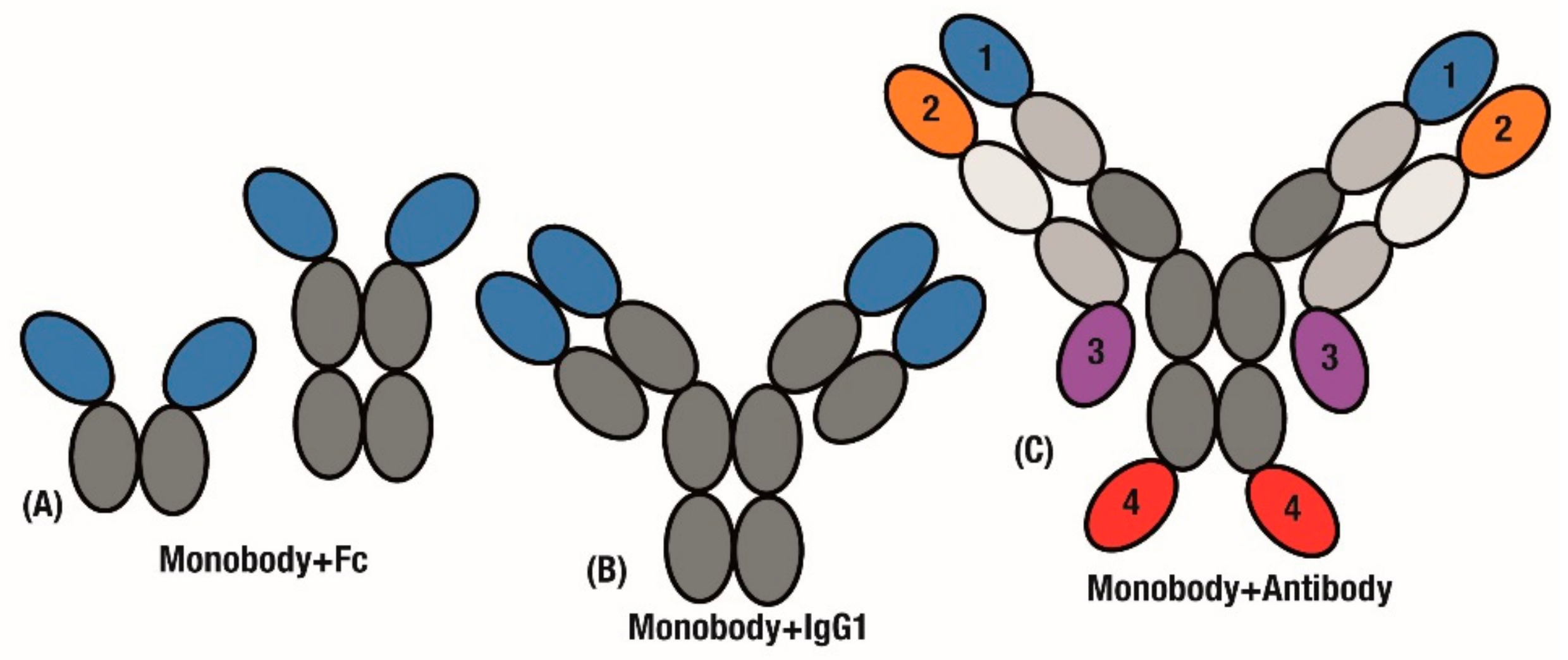
3.2. Combining Monobodies with Antibodies
3.3. Multi-Valent and Multi-Specific Monobodies
4. Future Applications of the Monobody Scaffold
4.1. Exploiting Responses to pH Change
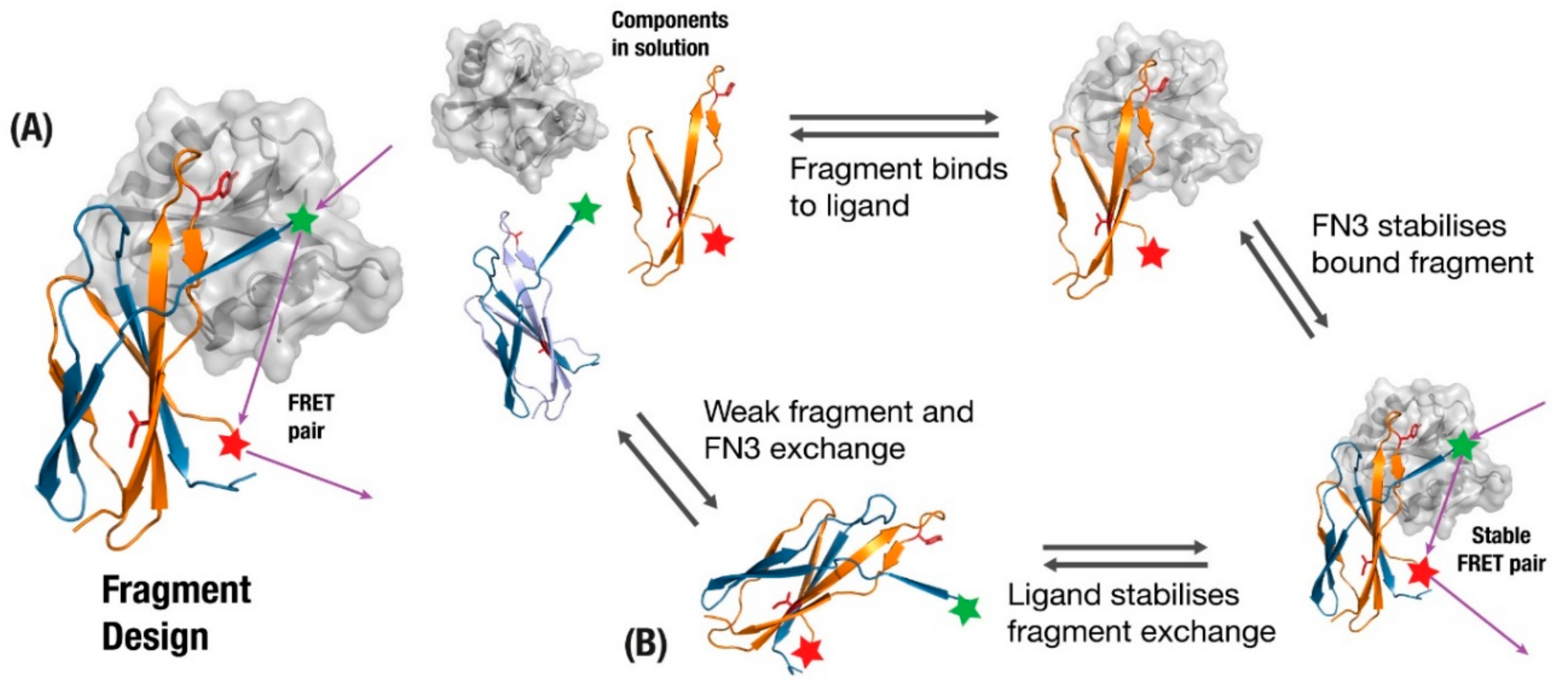
4.2. Native Strand-Exchange Function
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Vazquez-Lombardi, R.; Phan, T.G.; Zimmermann, C.; Lowe, D.; Jermutus, L.; Christ, D. Challenges and opportunities for non-antibody scaffold drugs. Drug Discov. Today 2015, 20, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Hantschel, O. Monobodies as possible next-generation protein therapeutics—A perspective. Swiss Med. Wkly. 2017, 147. [Google Scholar] [CrossRef]
- Sha, F.; Salzman, G.; Gupta, A.; Koide, S. Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci. 2017, 26, 910–924. [Google Scholar] [CrossRef] [PubMed]
- Koide, A.; Bailey, C.W.; Huang, X.; Koide, S. The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol. 1998, 284, 1141–1151. [Google Scholar] [CrossRef]
- Lipovsek, D. Adnectins: Engineered target-binding protein therapeutics. Protein Eng. Des. Sel. 2011, 24, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Mosher, D.F. Plasma fibronectin concentration: A risk factor for arterial thrombosis? Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1193–1195. [Google Scholar] [CrossRef]
- Binz, H.K.; Plückthun, A. Engineered proteins as specific binding reagents. Curr. Opin. Biotechnol. 2005, 16, 459–469. [Google Scholar] [CrossRef]
- Frejd, F.Y.; Kim, K.-T. Affibody molecules as engineered protein drugs. Exp. Mol. Med. 2017, 49, e306-e306. [Google Scholar] [CrossRef]
- Scott, C.J.; Taggart, C.C. Biologic protease inhibitors as novel therapeutic agents. Biochimie 2010, 92, 1681–1688. [Google Scholar] [CrossRef]
- Heinzelman, P.; Krais, J.; Ruben, E.; Pantazes, R. Engineering pH responsive fibronectin domains for biomedical applications. J. Biol. Eng. 2015, 9, 6. [Google Scholar] [CrossRef][Green Version]
- Koide, A.; Gilbreth, R.N.; Esaki, K.; Tereshko, V.; Koide, S. High-affinity single-domain binding proteins with a binary-code interface. Proc. Natl. Acad. Sci. USA 2007, 104, 6632–6637. [Google Scholar] [CrossRef] [PubMed]
- Hackel, B.J.; Ackerman, M.E.; Howland, S.W.; Wittrup, K.D. Stability and CDR Composition Biases Enrich Binder Functionality Landscapes. J. Mol. Biol. 2010, 401, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, V.P. Fibronectins: Structural-functional relationships. J. Evol. Biochem. Physiol. 2017, 53, 450–464. [Google Scholar] [CrossRef]
- Diem, M.D.; Hyun, L.; Yi, F.; Hippensteel, R.; Kuhar, E.; Lowenstein, C.; Swift, E.J.; O’Neil, K.T.; Jacobs, S.A.; Marx, J. Selection of high-affinity Centyrin FN3 domains from a simple library diversified at a combination of strand and loop positions. Protein Eng. Des. Sel. 2014, 27, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Ramamurthy, V.; Krystek, S.R.; Bush, A.; Wei, A.; Emanuel, S.L.; Das Gupta, R.; Janjua, A.; Cheng, L.; Murdock, M.; Abramczyk, B.; et al. Structures of Adnectin/Protein Complexes Reveal an Expanded Binding Footprint. Structure 2012, 20, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Koide, A.; Abbatiello, S.; Rothgery, L.; Koide, S. Probing protein conformational changes in living cells by using designer binding proteins: Application to the estrogen receptor. Proc. Natl. Acad. Sci. USA 2002, 99, 1253–1258. [Google Scholar] [CrossRef]
- Bloom, L.; Calabro, V. FN3: A new protein scaffold reaches the clinic. Drug Discov. Today 2009, 14, 949–955. [Google Scholar] [CrossRef]
- Lau, S.Y.; Siau, J.W.; Sobota, R.M.; Wang, C.I.; Zhong, P.; Lane, D.P.; Ghadessy, F.J. Synthetic 10FN3-based mono- and bivalent inhibitors of MDM2/X function. Protein Eng. Des. Sel. 2018, 31, 301–312. [Google Scholar] [CrossRef]
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of Fibronectin Extracellular Matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419. [Google Scholar] [CrossRef]
- Koide, A.; Wojcik, J.; Gilbreth, R.N.; Hoey, R.J.; Koide, S. Teaching an old scaffold new tricks: Monobodies constructed using alternative surfaces of the FN3 scaffold. J. Mol. Biol. 2012, 415, 393–405. [Google Scholar] [CrossRef]
- Zheng, H.; Bi, J.; Krendel, M.; Loh, S.N. Converting a binding protein into a biosensing conformational switch using protein fragment exchange. Biochemistry 2014, 53, 5505–5514. [Google Scholar] [CrossRef] [PubMed]
- Hober, S.; Lindbo, S.; Nilvebrant, J. Bispecific applications of non-immunoglobulin scaffold binders. Methods 2019, 154, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.J. Introduction to current and future protein therapeutics: A protein engineering perspective. Exp. Cell Res. 2011, 317, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Verdino, P.; Atwell, S.; Demarest, S.J. Emerging trends in bispecific antibody and scaffold protein therapeutics. Curr. Opin. Chem. Eng. 2018, 19, 107–123. [Google Scholar] [CrossRef]
- Leahy, D.J.; Aukhil, I.; Erickson, H.P. 2.0 Å Crystal Structure of a Four-Domain Segment of Human Fibronectin Encompassing the RGD Loop and Synergy Region. Cell 1996, 84, 155–164. [Google Scholar] [CrossRef]
- Koide, S.; Koide, A.; Lipovsek, D. Target-binding proteins based on the 10th human fibronectin type III domain (10Fn3). Methods Enzymol. 2012, 503, 135–156. [Google Scholar]
- Olson, C.A.; Roberts, R.W. Design, expression, and stability of a diverse protein library based on the human fibronectin type III domain. Protein Sci. 2007, 16, 476–484. [Google Scholar] [CrossRef]
- Olson, C.A.; Liao, H.I.; Sun, R.; Roberts, R.W. mRNA display selection of a high-affinity, modification-specific phospho-iκbα-binding fibronectin. ACS Chem. Biol. 2008, 3, 480–485. [Google Scholar] [CrossRef]
- Gross, G.G.; Junge, J.A.; Mora, R.J.; Kwon, H.-B.; Olson, C.A.; Takahashi, T.T.; Liman, E.R.; Ellis-Davies, G.C.R.; McGee, A.W.; Sabatini, B.L.; et al. Recombinant Probes for Visualizing Endogenous Synaptic Proteins in Living Neurons. Neuron 2013, 78, 971–985. [Google Scholar] [CrossRef]
- Nichols, A.L.; Noridomi, K.; Hughes, C.R.; Jalali-Yazdi, F.; Eaton, J.B.; Lai, L.H.; Advani, G.; Lukas, R.J.; Lester, H.A.; Chen, L.; et al. α1-FANGs: Protein Ligands Selective for the α-Bungarotoxin Site of the α1-Nicotinic Acetylcholine Receptor. ACS Chem. Biol. 2018, 13, 2568–2576. [Google Scholar] [CrossRef]
- Gilbreth, R.N.; Chacko, B.M.; Grinberg, L.; Swers, J.S.; Baca, M. Stabilization of the third fibronectin type III domain of human tenascin-C through minimal mutation and rational design. Protein Eng. Des. Sel. 2014, 27, 411–418. [Google Scholar] [CrossRef]
- Ng, S.P.; Billings, K.S.; Ohashi, T.; Allen, M.D.; Best, R.B.; Randles, L.G.; Erickson, H.P.; Clarke, J. Designing an extracellular matrix protein with enhanced mechanical stability. Proc. Natl. Acad. Sci. USA 2007, 104, 9633–9637. [Google Scholar] [CrossRef] [PubMed]
- Chandler, P.G.; Broendum, S.S.; Riley, B.T.; Spence, M.A.; Jackson, C.J.; McGowan, S.; Buckle, A.M. Strategies for Increasing Protein Stability. In Methods in Molecular Biology, Protein Nanotechnology, 3rd ed.; Gerrard, J., Domigan, L., Eds.; Humana: New York, NY, USA, 2020; Volume 2073, pp. 163–181. [Google Scholar]
- Jacobs, S.A.; Diem, M.D.; Luo, J.; Teplyakov, A.; Obmolova, G.; Malia, T.; Gilliland, G.L.; Oneil, K.T. Design of novel FN3 domains with high stability by a consensus sequence approach. Protein Eng. Des. Sel. 2012, 25, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.D.; Cardoso, R.M.F.; Lin, T.; Spinka-Doms, T.; Klein, D.; Jacobs, S.A.; Dudkin, V.; Gilliland, G.; O’Neil, K.T. Engineering a targeted delivery platform using Centyrins. Protein Eng. Des. Sel. 2016, 29, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Porebski, B.T.; Nickson, A.A.; Hoke, D.E.; Hunter, M.R.; Zhu, L.; McGowan, S.; Webb, G.I.; Buckle, A.M. Structural and dynamic properties that govern the stability of an engineered fibronectin type III domain. Protein Eng. Des. Sel. 2015, 28, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, M.; Skerra, A. Engineered protein scaffolds as next-generation antibody therapeutics. Curr. Opin. Chem. Biol. 2009, 13, 245–255. [Google Scholar] [CrossRef]
- Chen, T.F.; de Picciotto, S.; Hackel, B.J.; Wittrup, K.D. Engineering Fibronectin-Based Binding Proteins by Yeast Surface Display. In Methods in Enzymology, Methods in Protein Design, 1st ed.; Keating, A., Ed.; Elsevier Inc.: San Diego, CA, USA, 2013; Volume 523, pp. 303–326. ISBN 9780123942920. [Google Scholar]
- Gorman, K.; McGinnis, J.; Kay, B. Generating FN3-Based Affinity Reagents through Phage Display. Curr. Protoc. Chem. Biol. 2018, 10, e39. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Aha, P.; Gu, K.; Kuimelis, R.G.; Kurz, M.; Lam, T.; Lim, A.C.; Liu, H.; Lohse, P.A.; Sun, L.; et al. Directed Evolution of High-Affinity Antibody Mimics Using mRNA Display. Chem. Biol. 2002, 9, 933–942. [Google Scholar] [CrossRef]
- Liu, F.; Su, L.; Chen, Z.; Feng, D.; Wei, J.; Sun, J. Construction of small molecular CTLA4 analogs with CD80-binding affinity. Biochem. Biophys. Res. Commun. 2019, 513, 694–700. [Google Scholar] [CrossRef]
- Hussain, M.; Angus, S.P.; Kuhlman, B. Engineering a Protein Binder Specific for p38α with Interface Expansion. Biochemistry 2018, 57, 4526–4535. [Google Scholar] [CrossRef]
- Lewis, S.M.; Kuhlman, B.A. Anchored design of protein-protein interfaces. PLoS ONE 2011, 6, e20872. [Google Scholar] [CrossRef] [PubMed]
- Ewert, S.; Honegger, A.; Plückthun, A. Stability improvement of antibodies for extracellular and intracellular applications: CDR grafting to stable frameworks and structure-based framework engineering. Methods 2004, 34, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Verhoeyen, M.; Milstein, C.; Winter, G. Reshaping human antibodies: Grafting an antilysozyme activity. Science 1988, 239, 1534–1536. [Google Scholar] [CrossRef]
- Porebski, B.T.; Conroy, P.J.; Drinkwater, N.; Schofield, P.; Vazquez-Lombardi, R.; Hunter, M.R.; Hoke, D.E.; Christ, D.; McGowan, S.; Buckle, A.M. Circumventing the stability-function trade-off in an engineered FN3 domain. Protein Eng. Des. Sel. 2016, 29, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Mamluk, R.; Carvajal, I.M.; Morse, B.A.; Wong, H.K.; Abramowitz, J.; Aslanian, S.; Lim, A.-C.; Gokemeijer, J.; Storek, M.J.; Lee, J.; et al. Anti-tumor effect of CT-322 as an Adnectin inhibitor of vascular endothelial growth factor receptor-2. mAbs 2010, 2, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Schiff, D.; Kesari, S.; De Groot, J.; Mikkelsen, T.; Drappatz, J.; Coyle, T.; Fichtel, L.; Silver, B.; Walters, I.; Reardon, D. Phase 2 study of CT-322, a targeted biologic inhibitor of VEGFR-2 based on a domain of human fibronectin, in recurrent glioblastoma. Investig. New Drugs 2015, 33, 247–253. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Lambrechts, D.; Prenen, H.; Jain, R.K.; Carmeliet, P. Lessons from the adjuvant bevacizumab trial on colon cancer: What next? J. Clin. Oncol. 2011, 29, 1–4. [Google Scholar] [CrossRef]
- Aghaabdollahian, S.; Ahangari Cohan, R.; Norouzian, D.; Davami, F.; Asadi Karam, M.R.; Torkashvand, F.; Vaseghi, G.; Moazzami, R.; Latif Dizaji, S. Enhancing bioactivity, physicochemical, and pharmacokinetic properties of a nano-sized, anti-VEGFR2 Adnectin, through PASylation technology. Sci. Rep. 2019, 9, 2978. [Google Scholar] [CrossRef]
- Ding, N.; Fu, X.; Ruan, Y.; Zhu, J.; Guo, P.; Han, L.; Zhang, J.; Hu, X. Extracellular production of recombinant N-glycosylated anti-VEGFR2 monobody in leaky Escherichia coli strain. Biotechnol. Lett. 2019, 41, 1265–1274. [Google Scholar] [CrossRef]
- Abou-Elkacem, L.; Wilson, K.E.; Johnson, S.M.; Chowdhury, S.M.; Bachawal, S.; Hackel, B.J.; Tian, L.; Willmann, J.K. Ultrasound molecular imaging of the breast cancer neovasculature using engineered fibronectin scaffold ligands: A novel class of targeted contrast ultrasound agent. Theranostics 2016, 6, 1740–1752. [Google Scholar] [CrossRef]
- Kulemzin, S.V.; Gorchakov, A.A.; Chikaev, A.N.; Kuznetsova, V.V.; Volkova, O.Y.; Matvienko, D.A.; Petukhov, A.V.; Zaritskey, A.Y.; Taranin, A.V. VEGFR2-specific FnCAR effectively redirects the cytotoxic activity of T cells and YT NK cells. Oncotarget 2018, 9, 9021. [Google Scholar] [CrossRef] [PubMed]
- Kükenshöner, T.; Schmit, N.E.; Bouda, E.; Sha, F.; Pojer, F.; Koide, A.; Seeliger, M.; Koide, S.; Hantschel, O. Selective Targeting of SH2 Domain–Phosphotyrosine Interactions of Src Family Tyrosine Kinases with Monobodies. J. Mol. Biol. 2017, 429, 1364–1380. [Google Scholar] [CrossRef] [PubMed]
- Zorba, A.; Nguyen, V.; Koide, A.; Hoemberger, M.; Zheng, Y.; Kutter, S.; Kim, C.; Koide, S.; Kern, D. Allosteric modulation of a human protein kinase with monobodies. Proc. Natl. Acad. Sci. USA 2019, 116, 201906024. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, B.C.; Newstead, S.; Stockbridge, R.B. Cork-in-Bottle Occlusion of Fluoride Ion Channels by Crystallization Chaperones. Structure 2018, 26, 635–639. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Lipovšek, D.; Carvajal, I.; Allentoff, A.J.; Barros, A.; Brailsford, J.; Cong, Q.; Cotter, P.; Gangwar, S.; Hollander, C.; Lafont, V.; et al. Adnectin–drug conjugates for Glypican-3-specific delivery of a cytotoxic payload to tumors. Protein Eng. Des. Sel. 2018, 31, 159–171. [Google Scholar] [CrossRef]
- Deonarain, M.; Yahioglu, G.; Stamati, I.; Pomowski, A.; Clarke, J.; Edwards, B.; Diez-Posada, S.; Stewart, A. Small-Format Drug Conjugates: A Viable Alternative to ADCs for Solid Tumours? Antibodies 2018, 7, 16. [Google Scholar] [CrossRef]
- Shi, C.; Goldberg, S.; Lin, T.; Dudkin, V.; Widdison, W.; Harris, L.; Wilhelm, S.; Jmeian, Y.; Davis, D.; O’Neil, K.; et al. Bioanalytical workflow for novel scaffold protein-drug conjugates: Quantitation of total Centyrin protein, conjugated Centyrin and free payload for Centyrin-drug conjugate in plasma and tissue samples using liquid chromatography-tandem mass spectrometry. Bioanalysis 2018, 10, 1651–1665. [Google Scholar] [CrossRef]
- Shi, C.; Goldberg, S.; Lin, T.; Dudkin, V.; Widdison, W.; Harris, L.; Wilhelm, S.; Jmeian, Y.; Davis, D.; O’Neil, K.; et al. LC/MS/MS Bioanalysis of Protein-Drug Conjugates—The Importance of Incorporating Succinimide Hydrolysis Products. Anal. Chem. 2018, 90, 5314–5321. [Google Scholar] [CrossRef]
- Natarajan, A.; Patel, C.B.; Ramakrishnan, S.; Panesar, P.S.; Long, S.R.; Gambhir, S.S. A Novel Engineered Small Protein for Positron Emission Tomography Imaging of Human Programmed Death Ligand-1: Validation in Mouse Models and Human Cancer Tissues. Clin. Cancer Res. 2019, 25, 1774–1785. [Google Scholar] [CrossRef]
- Schmit, N.E.; Neopane, K.; Hantschel, O. Targeted Protein Degradation through Cytosolic Delivery of Monobody Binders Using Bacterial Toxins. ACS Chem. Biol. 2019, 14, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Cetin, M.; Evenson, W.E.; Gross, G.G.; Jalali-Yazdi, F.; Krieger, D.; Arnold, D.; Takahashi, T.T.; Roberts, R.W. RasIns: Genetically Encoded Intrabodies of Activated Ras Proteins. J. Mol. Biol. 2017, 429, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.; Natarajan, A.; Chan, C.T.; Panesar, P.S.; Gambhir, S.S. Engineering of a novel subnanomolar affinity fibronectin III domain binder targeting human programmed death-ligand 1. Protein Eng. Des. Sel. 2019. [Google Scholar] [CrossRef] [PubMed]
- Broos, K.; Lecocq, Q.; Raes, G.; Devoogdt, N.; Keyaerts, M.; Breckpot, K. Noninvasive imaging of the PD-1: PD-L1 immune checkpoint: Embracing nuclear medicine for the benefit of personalized immunotherapy. Theranostics 2018, 8, 3559–3570. [Google Scholar] [CrossRef]
- Donnelly, D.J.; Smith, R.A.; Morin, P.; Lipovšek, D.; Gokemeijer, J.; Cohen, D.; Lafont, V.; Tran, T.; Cole, E.L.; Wright, M.; et al. Synthesis and Biologic Evaluation of a Novel 18 F-Labeled Adnectin as a PET Radioligand for Imaging PD-L1 Expression. J. Nucl. Med. 2018, 59, 529–535. [Google Scholar] [CrossRef]
- Leung, D.K.; De Langen, J.; Raunig, D.; Niemeijer, A.-L.N.; Smit, E.F.; Boellaard, R.; Vallez-Garcia, D.; van Dongen, G.a.M.S.; Windhorst, A.D.; Huisman, M.C.; et al. Whole body PD-L1 PET in patients with NSCLC and melanoma. J. Clin. Oncol. 2018, 36, 139. [Google Scholar] [CrossRef]
- Natarajan, A.; Abou-Elkacem, L. FN3 Protein Conjugates for Cancer Diagnosis and Imaging Studies. In Methods in Molecular Biology, Bioconjugation; Massa, S., Devoogdt, N., Eds.; Humana: New York, NY, USA, 2019; Volume 2033, pp. 301–313. [Google Scholar]
- Mahalingam, S.M.; Dudkin, V.Y.; Goldberg, S.; Klein, D.; Yi, F.; Singhal, S.; O’Neil, K.T.; Low, P.S. Evaluation of a Centyrin-Based Near-Infrared Probe for Fluorescence-Guided Surgery of Epidermal Growth Factor Receptor Positive Tumors. Bioconjugate Chem. 2017, 28, 2865–2873. [Google Scholar] [CrossRef]
- Yeh, J.T.H.; Binari, R.; Gocha, T.; Dasgupta, R.; Perrimon, N. PAPTi: A peptide aptamer interference toolkit for perturbation of protein-protein interaction networks. Sci. Rep. 2013, 3, 1–8. [Google Scholar] [CrossRef]
- Gupta, A.; Xu, J.; Lee, S.; Tsai, S.T.; Zhou, B.; Kurosawa, K.; Werner, M.S.; Koide, A.; Ruthenburg, A.J.; Dou, Y.; et al. Facile target validation in an animal model with intracellularly expressed monobodies. Nat. Chem. Biol. 2018, 14, 895–900. [Google Scholar] [CrossRef]
- Ludwicki, M.B.; Li, J.; Stephens, E.A.; Roberts, R.W.; Koide, S.; Hammond, P.T.; Delisa, M.P. Broad-Spectrum Proteome Editing with an Engineered Bacterial Ubiquitin Ligase Mimic. ACS Cent. Sci. 2019, 5, 852–866. [Google Scholar] [CrossRef]
- Fulcher, L.J.; Hutchinson, L.D.; Macartney, T.J.; Turnbull, C.; Sapkota, G.P. Targeting endogenous proteins for degradation through the affinity-directed protein missile system. Open Biol. 2017, 7, 170066. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.G.; Thompson, T.B. Fibronectin-based scaffold domain proteins that bind myostatin: A patent evaluation of WO2014043344. Expert Opin. Ther. Pat. 2015, 25, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Gapizov, S.S.; Petrovskaya, L.E.; Shingarova, L.N.; Kryukova, E.A.; Boldyreva, E.F.; Lukashev, E.P.; Yakimov, S.A.; Svirshchevskaya, E.V.; Dolgikh, D.A.; Kirpichnikov, M.P. Fusion with an albumin-binding domain improves pharmacokinetics of an αvβ3-integrin binding fibronectin scaffold protein. Biotechnol. Appl. Biochem. 2019, 66, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.A.; Gibbs, A.C.; Conk, M.; Yi, F.; Maguire, D.; Kane, C.; O’neil, K.T. Fusion to a highly stable consensus albumin binding domain allows for tunable pharmacokinetics. Protein Eng. Des. Sel. 2015, 28, 385–393. [Google Scholar] [CrossRef]
- Chan, R.; Buckley, P.T.; O’Malley, A.; Sause, W.E.; Alonzo, F.; Lubkin, A.; Boguslawski, K.M.; Payne, A.; Fernandez, J.; Strohl, W.R.; et al. Identification of biologic agents to neutralize the bicomponent leukocidins of Staphylococcus aureus. Sci. Transl. Med. 2019, 11, eaat0882. [Google Scholar] [CrossRef]
- Seidah, N.G.; Prat, A.; Pirillo, A.; Catapano, A.L.; Norata, G.D. Novel strategies to target proprotein convertase subtilisin kexin 9: Beyond monoclonal antibodies. Cardiovasc. Res. 2019, 115, 510–518. [Google Scholar] [CrossRef]
- Kazi, D.S.; Penko, J.; Coxson, P.G.; Moran, A.E.; Ollendorf, D.A.; Tice, J.A.; Bibbins-Domingo, K. Updated cost-effectiveness analysis of PCSK9 inhibitors based on the results of the FOURIER trial. JAMA 2017, 318, 748–750. [Google Scholar] [CrossRef]
- Mitchell, T.; Chao, G.; Sitkoff, D.; Lo, F.; Monshizadegan, H.; Meyers, D.; Low, S.; Russo, K.; DiBella, R.; Denhez, F.; et al. Pharmacologic Profile of the Adnectin BMS-962476, a Small Protein Biologic Alternative to PCSK9 Antibodies for Low-Density Lipoprotein Lowering. J. Pharmacol. Exp. Ther. 2014, 350, 412–424. [Google Scholar] [CrossRef]
- Stein, E.; Biernat, L.; Turner, T.; Lee, J.; Arumugam, U.; Kasichayanula, S.; Kranz, T. Ldl Cholesterol Reduction With Bms-962476, an Adnectin Inhibitor of Pcsk9: Results of a Single Ascending Dose Study. J. Am. Coll. Cardiol. 2014, 63, A1372. [Google Scholar] [CrossRef]
- Masuda, Y.; Yamaguchi, S.; Suzuki, C.; Aburatani, T.; Nagano, Y.; Miyauchi, R.; Suzuki, E.; Yamamura, N.; Nagatomo, K.; Ishihara, H.; et al. Generation and Characterization of a Novel Small Biologic Alternative to Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Antibodies, DS-9001a, Albumin Binding Domain–Fused Anticalin Protein. J. Pharmacol. Exp. Ther. 2018, 365, 368–378. [Google Scholar] [CrossRef]
- Stein, E.; Turner, T.; Biernat, L.; Dimova, D.; Zhou, R.; Dai, M.; Daniels, C.; Mitchell, T. Low Density Lipoprotein Cholesterol Reduction and Safety With Lib003, an Anti-Proprotein Convertase Subtilisin/Kexin Type 9 Fusion Protein: Results of a Randomized, Double-Blind, Placebo-Controlled, Single Ascending Dose Study. J. Am. Coll. Cardiol. 2019, 73, 1714. [Google Scholar] [CrossRef]
- Stein, E.; Toth, P.; Butcher, M.; Kereiakes, D.; Magnu, P.; Bays, H.; Zhou, R.; Turner, T.A. Safety, Tolerability And Ldl-C Reduction With A Novel Anti-Pcsk9 Recombinant Fusion Protein (Lib003): Results Of A Randomized, Double-Blind, Placebo-Controlled, Phase 2 Study. Atherosclerosis 2019, 287, e7. [Google Scholar] [CrossRef]
- Hanna, M.G.; Badrising, U.A.; Benveniste, O.; Lloyd, T.E.; Needham, M.; Chinoy, H.; Aoki, M.; Machado, P.M.; Liang, C.; Reardon, K.A.; et al. Safety and efficacy of intravenous bimagrumab in inclusion body myositis (RESILIENT): A randomised, double-blind, placebo-controlled phase 2b trial. Lancet Neurol. 2019, 18, 834–844. [Google Scholar] [CrossRef]
- Zhu, Y.; D’Arienzo, C.; Lou, Z.; Kozhich, A.; Madireddi, M.; Chimalakonda, A.; Tymiak, A.; Olah, T.V. LC-MS/MS multiplexed assay for the quantitation of a therapeutic protein BMS-986089 and the target protein Myostatin. Bioanalysis 2016, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Madireddi, M.; Malone, H.; Kukral, D.; Chimalakonda, A.; Kozhich, A.; Xiling, Y.; Swain, J.; Yamniuk, A.; Ahlijanian, M. BMS-986089 is a high affinity anti-myostatin adnectin that increases muscle volume in three preclinical species. Neuromuscul. Disord. 2016, 26, S94–S95. [Google Scholar] [CrossRef]
- Jacobsen, L.; Bechtold, C.; Tirucherai, G.; Ahlijanian, M.; Luo, F. BMS-986089: A novel adnectin protein that dose dependently lowers free myostatin and increases muscle volume and lean body mass. Neuromuscul. Disord. 2016, 26, S95. [Google Scholar] [CrossRef]
- Wagner, K.R.; Wong, B.L.; Byrne, B.J.; Tian, C.; Jacobsen, L.K.; Tirucherai, G.S.; Rabbia, M.; Kletzl, H.; Dukart, J.; Ong, R.; et al. A Phase 1b/2 Study of the Anti-Myostatin Adnectin RG6206 (BMS-986089) in Ambulatory Boys with Duchenne Muscular Dystrophy: A 72-Week Treatment Update (P1.6-062). In Proceedings of the 71st Annual Meeting of the American-Academy-of-Neurology (AAN), Philadelphia, PA, USA, 4–10 May 2019; Volume 92 (Suppl. 15). Abstract P1.6-062. [Google Scholar]
- Swers, J.S.; Grinberg, L.; Wang, L.; Feng, H.; Lekstrom, K.; Carrasco, R.; Xiao, Z.; Inigo, I.; Leow, C.C.; Wu, H.; et al. Multivalent scaffold proteins as superagonists of TRAIL receptor 2-induced apoptosis. Mol. Cancer Ther. 2013, 12, 1235–1244. [Google Scholar] [CrossRef]
- Suurs, F.V.; Lub-de Hooge, M.N.; de Vries, E.G.E.; de Groot, D.J.A. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol. Ther. 2019, 201, 103–119. [Google Scholar] [CrossRef]
- Zhang, D.; Whitaker, B.; Derebe, M.G.; Chiu, M.L. FcγRII-binding Centyrins mediate agonism and antibody-dependent cellular phagocytosis when fused to an anti-OX40 antibody. mAbs 2018, 10, 463–475. [Google Scholar] [CrossRef]
- Han, X.; Cinay, G.E.; Zhao, Y.; Guo, Y.; Zhang, X.; Wang, P. Adnectin-Based Design of Chimeric Antigen Receptor for T Cell Engineering. Mol. Ther. 2017, 25, 2466–2476. [Google Scholar] [CrossRef]
- Hermanson, D.L.; Barnett, B.E.; Rengarajan, S.; Codde, R.; Wang, X.; Tan, Y.; Martin, C.E.; Smith, J.B.; He, J.; Mathur, R.; et al. A Novel Bcma-Specific, Centyrin-Based CAR-T Product for the Treatment of Multiple Myeloma. Blood 2016, 128, 2127. [Google Scholar] [CrossRef]
- Smith, J.B.; Codde, R.; Tan, Y.; Barnett, B.E.; Hermanson, D.; Rengarajan, S.; Ostertag, E.M.; Shedlock, D.J. Abstract A071: PSMA-specific CARTyrin T-stem cell memory therapy eliminates solid tumor in subcutaneous prostate cancer model. In Proceedings of the AACR Special Conference on Prostate Cancer - Advances in Basic, Translational, and Clinical Research, Orlando, FL, USA, 2–5 December 2017; Abstract A071. pp. 62–63. [Google Scholar]
- Gregory, T.K.; Berdeja, J.G.; Patel, K.K.; Ali, S.A.; Cohen, A.D.; Costello, C.; Ostertag, E.M.; de Silva, N.; Shedlock, D.J.; Resler, M.; et al. Abstract CT130: Clinical trial of P-BCMA-101 T stem cell memory (Tscm) CAR-T cells in relapsed/refractory (r/r) multiple myeloma (MM). In Proceedings of the Annual Meeting of the American-Association-for-Cancer-Research (AACR), Chicago, IL, USA, 14–18 April 2018. Abstract CT130. [Google Scholar]
- Costello, C.L.; Gregory, T.K.; Ali, S.A.; Berdeja, J.G.; Patel, K.K.; Shah, N.D.; Ostertag, E.; Martin, C.; Ghoddusi, M.; Shedlock, D.J.; et al. Phase 2 Study of the Response and Safety of P-Bcma-101 CAR-T Cells in Patients with Relapsed/Refractory (r/r) Multiple Myeloma (MM) (PRIME). Blood 2019, 134, 3184-3184. [Google Scholar] [CrossRef]
- Luthra, A.; Langley, D.B.; Schofield, P.; Jackson, J.; Abdelatti, M.; Rouet, R.; Nevoltris, D.; Mazigi, O.; Crossett, B.; Christie, M.; et al. Human Antibody Bispecifics through Phage Display Selection. Biochemistry 2019, 58, 1701–1704. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Morse, B.A.; Delventhal, V.; Carvajal, I.M.; Konerding, M.A. Anti-VEGFR2 and anti-IGF-1R-Adnectins inhibit Ewing’s sarcoma A673-xenograft growth and normalize tumor vascular architecture. Angiogenesis 2012, 15, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, S.L.; Engle, L.J.; Chao, G.; Zhu, R.-R.; Cao, C.; Lin, Z.; Yamniuk, A.P.; Hosbach, J.; Brown, J.; Fitzpatrick, E.; et al. A fibronectin scaffold approach to bispecific inhibitors of epidermal growth factor receptor and insulin-like growth factor-I receptor. mAbs 2011, 3, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Wensel, D.; Sun, Y.; Li, Z.; Zhang, S.; Picarillo, C.; McDonagh, T.; Fabrizio, D.; Cockett, M.; Krystal, M.; Davis, J. Discovery and Characterization of a Novel CD4-Binding Adnectin with Potent Anti-HIV Activity. Antimicrob. Agents Chemother. 2017, 61, e00508-17. [Google Scholar] [CrossRef]
- Wensel, D.; Sun, Y.; Davis, J.; Li, Z.; Zhang, S.; McDonagh, T.; Fabrizio, D.; Cockett, M.; Krystal, M. A Novel gp41-Binding Adnectin with Potent Anti-HIV Activity Is Highly Synergistic when Linked to a CD4-Binding Adnectin. J. Virol. 2018, 92, e00421-18. [Google Scholar] [CrossRef]
- Cuesta, Á.M.; Sainz-Pastor, N.; Bonet, J.; Oliva, B.; Álvarez-Vallina, L. Multivalent antibodies: When design surpasses evolution. Trends Biotechnol. 2010, 28, 355–362. [Google Scholar] [CrossRef]
- Karnell, J.L.; Albulescu, M.; Drabic, S.; Wang, L.; Moate, R.; Baca, M.; Oganesyan, V.; Gunsior, M.; Thisted, T.; Yan, L.; et al. A CD40L-targeting protein reduces autoantibodies and improves disease activity in patients with autoimmunity. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Oganesyan, V.; Ferguson, A.; Grinberg, L.; Wang, L.; Phipps, S.; Chacko, B.; Drabic, S.; Thisted, T.; Baca, M. Fibronectin type III domains engineered to bind CD40L: Cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis of two complexes. Acta Crystallogr. Sect. F 2013, 69, 1045–1048. [Google Scholar] [CrossRef]
- Coyle, A.; Baca, M.; Thisted, T.; Drabic, S.; Grinberg, L.; Novarra, S.; Oganesyan, V.; Herbst, R.; Spencer, D.K. CD40L-Specific TN3-Derived Scaffolds and Methods of Use Thereof (57) WO 2013/055745 A2 2013. U.S. Patent No. 10,000,553, 18 April 2013. [Google Scholar]
- Albulescu, M.; Gunsior, M.; Li, J.; Bush, J.; Godwood, A.; Miday, R.; Grant, E.; Howe, D.; Faggioni, R.; Roskos, L. SAT0249 Safety, pharmacokinetics, pharmacodynamics and inhibition of T-cell dependent antibody response (TDAR) with MEDI4920, a novel, engineered CD40 ligand (CD40L) antagonist: Results of a first-time-in-human study. In Proceedings of the Poster Presentations; BMJ Publishing Group Ltd.: London, UK; European League Against Rheumatism: Zurich, Switzerland, 2017; pp. 867–868. [Google Scholar]
- Nicholson, S.M.; Casey, K.A.; Gunsior, M.; Drabic, S.; Iverson, W.; Cook, H.; Scott, S.; O’Day, T.; Karanth, S.; Dixit, R.; et al. The enhanced immunopharmacology of VIB4920, a novel Tn3 fusion protein and CD40L antagonist, and assessment of its safety profile in cynomolgus monkeys. Br. J. Pharmacol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Greer, Y.E.; Gilbert, S.F.; Gril, B.; Narwal, R.; Peacock Brooks, D.L.; Tice, D.A.; Steeg, P.S.; Lipkowitz, S. MEDI3039, a novel highly potent tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptor 2 agonist, causes regression of orthotopic tumors and inhibits outgrowth of metastatic triple-negative breast cancer. Breast Cancer Res. 2019, 21, 27. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Wu, J.; Valencia, C.A.; Liu, R. Fibronectin Type III Domain Based Monobody with High Avidity. Biochemistry 2007, 46, 12656–12664. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.; Gao, M.; Schulten, K.; Vogel, V. Tuning the Mechanical Stability of Fibronectin Type III Modules through Sequence Variations. Structure 2004, 12, 21–30. [Google Scholar] [CrossRef]
- Ingham, K.C.; Brew, S.A.; Huff, S.; Litvinovich, S.V. Cryptic self-association sites in type III modules of fibronectin. J. Biol. Chem. 1997, 272, 1718–1724. [Google Scholar] [CrossRef] [PubMed]
- Jain, T.; Sun, T.; Durand, S.; Hall, A.; Houston, N.R.; Nett, J.H.; Sharkey, B.; Bobrowicz, B.; Caffry, I.; Yu, Y.; et al. Biophysical properties of the clinical-stage antibody landscape. Proc. Natl. Acad. Sci. USA 2017, 114, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Teplyakov, A.; Obmolova, G.; Malia, T.; Wu, S.J.; Beil, E.; Baker, A.; Swencki-Underwood, B.; Zhao, Y.; Sprenkle, J.; et al. Structure of the EMMPRIN N-terminal domain 1: Dimerization via β-strand swapping. Proteins Struct. Funct. Bioinform. 2009, 77, 1009–1014. [Google Scholar] [CrossRef]
- Spinelli, S.; Desmyter, A.; Frenken, L.; Verrips, T.; Tegoni, M.; Cambillau, C. Domain swapping of a llama VHH domain builds a crystal-wide β-sheet structure. FEBS Lett. 2004, 564, 35–40. [Google Scholar] [CrossRef]
- Ultsch, M.H.; Wiesmann, C.; Simmons, L.C.; Henrich, J.; Yang, M.; Reilly, D.; Bass, S.H.; De Vos, A.M. Crystal structures of the neurotrophin-binding domain of TrkA, TrkB and TrkC. J. Mol. Biol. 1999, 290, 149–159. [Google Scholar] [CrossRef]
- Gabrielson, J.P.; Weiss, W.F. Technical Decision-Making with Higher Order Structure Data: Starting a New Dialogue. J. Pharm. Sci. 2015, 104, 1240–1245. [Google Scholar] [CrossRef]
- Orphanou, C.; Gervais, D. Higher-order structure and conformational change in biopharmaceuticals. J. Chem. Technol. Biotechnol. 2018, 93, 2477–2485. [Google Scholar] [CrossRef]
- Teplyakov, A.; Obmolova, G.; Malia, T.J.; Luo, J.; Jacobs, S.A.; Chan, W.; Domingo, D.; Baker, A.; O’Neil, K.T.; Gilliland, G.L. C-terminal β-strand swapping in a consensus-derived fibronectin Type III scaffold. Proteins Struct. Funct. Bioinform. 2014, 82, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Teplyakov, A.; Obmolova, G.; Malia, T.J.; Chan, W.; Jacobs, S.A.; O’Neil, K.T.; Gilliland, G.L. N-terminal β-strand swapping in a consensus-derived alternative scaffold driven by stabilizing hydrophobic interactions. Proteins Struct. Funct. Bioinform. 2014, 82, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; O’Neil, S.R.; Lipovsek, D.; Chen, G. Conformational Assessment of Adnectin and Adnectin-Drug Conjugate by Hydrogen/Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2018, 29, 1524–1531. [Google Scholar] [CrossRef]
- Hackel, B.J.; Kapila, A.; Dane Wittrup, K. Picomolar Affinity Fibronectin Domains Engineered Utilizing Loop Length Diversity, Recursive Mutagenesis, and Loop Shuffling. J. Mol. Biol. 2008, 381, 1238–1252. [Google Scholar] [CrossRef]
- Rousseau, F.; Schymkowitz, J.; Itzhaki, L.S. Implications of 3D domain swapping for protein folding, misfolding and function. Adv. Exp. Med. Biol. 2012, 747, 137–152. [Google Scholar]
- Trainor, K.; Gingras, Z.; Shillingford, C.; Malakian, H.; Gosselin, M.; Lipovsek, D.; Meiering, E.M. Ensemble Modeling and Intracellular Aggregation of an Engineered Immunoglobulin-Like Domain. J. Mol. Biol. 2016, 428, 1365–1374. [Google Scholar] [CrossRef]
- Ito, H.O.; Soutome, S.; Inoue, M. Inhibition of fibronectin binding of some bacterial cells by subtle pH increase within the physiological range. J. Microbiol. Methods 2003, 55, 29–34. [Google Scholar] [CrossRef]
- Chaparro-Riggers, J.; Liang, H.; DeVay, R.M.; Bai, L.; Sutton, J.E.; Chen, W.; Geng, T.; Lindquist, K.; Casas, M.G.; Boustany, L.M.; et al. Increasing serum half-life and extending cholesterol lowering in vivo by engineering antibody with pH-sensitive binding to PCSK9. J. Biol. Chem. 2012, 287, 11090–11097. [Google Scholar] [CrossRef]
- Lee, C.H.; Kang, T.H.; Godon, O.; Watanabe, M.; Delidakis, G.; Gillis, C.M.; Sterlin, D.; Hardy, D.; Cogné, M.; Macdonald, L.E.; et al. An engineered human Fc domain that behaves like a pH-toggle switch for ultra-long circulation persistence. Nat. Commun. 2019, 10, 1–11. [Google Scholar]
- Ramírez-García, P.D.; Retamal, J.S.; Shenoy, P.; Imlach, W.; Sykes, M.; Truong, N.; Constandil, L.; Pelissier, T.; Nowell, C.J.; Khor, S.Y.; et al. A pH-responsive nanoparticle targets the neurokinin 1 receptor in endosomes to prevent chronic pain. Nat. Nanotechnol. 2019, 14, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Koide, S. Rational conversion of affinity reagents into label-free sensors for peptide motifs by designed allostery. ACS Chem. Biol. 2010, 5, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Islam, J.; Riley, B.T.; Fercher, C.; Jones, M.L.; Buckle, A.M.; Howard, C.B.; Cox, R.P.; Bell, T.D.M.; Mahler, S.; Corrie, S.R. Wavelength-Dependent Fluorescent Immunosensors via Incorporation of Polarity Indicators near the Binding Interface of Antibody Fragments. Anal. Chem. 2019, 91, 7631–7638. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.; Loh, S.N. Construction of Allosteric Protein Switches by Alternate Frame Folding and Intermolecular Fragment Exchange. In Synthetic Protein Switches: Methods and Protocols; Science and Business Media: Berlin/Heidelberg, Germany, 2017; Volume 1596, pp. 27–41. ISBN 978-1-4939-6938-8. [Google Scholar]
- Arnold, F.H. Design by Directed Evolution. Acc. Chem. Res. 1998, 31, 125–131. [Google Scholar] [CrossRef]
- Winter, G.; Griffiths, A.D.; Hawkins, R.E.; Hoogenboom, H.R. Making Antibodies by Phage Display Technology. Annu. Rev. Immunol. 1994, 12, 433–455. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandler, P.G.; Buckle, A.M. Development and Differentiation in Monobodies Based on the Fibronectin Type 3 Domain. Cells 2020, 9, 610. https://doi.org/10.3390/cells9030610
Chandler PG, Buckle AM. Development and Differentiation in Monobodies Based on the Fibronectin Type 3 Domain. Cells. 2020; 9(3):610. https://doi.org/10.3390/cells9030610
Chicago/Turabian StyleChandler, Peter G., and Ashley M. Buckle. 2020. "Development and Differentiation in Monobodies Based on the Fibronectin Type 3 Domain" Cells 9, no. 3: 610. https://doi.org/10.3390/cells9030610
APA StyleChandler, P. G., & Buckle, A. M. (2020). Development and Differentiation in Monobodies Based on the Fibronectin Type 3 Domain. Cells, 9(3), 610. https://doi.org/10.3390/cells9030610