Moderate Exercise Modulates Tumor Metabolism of Triple-Negative Breast Cancer

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
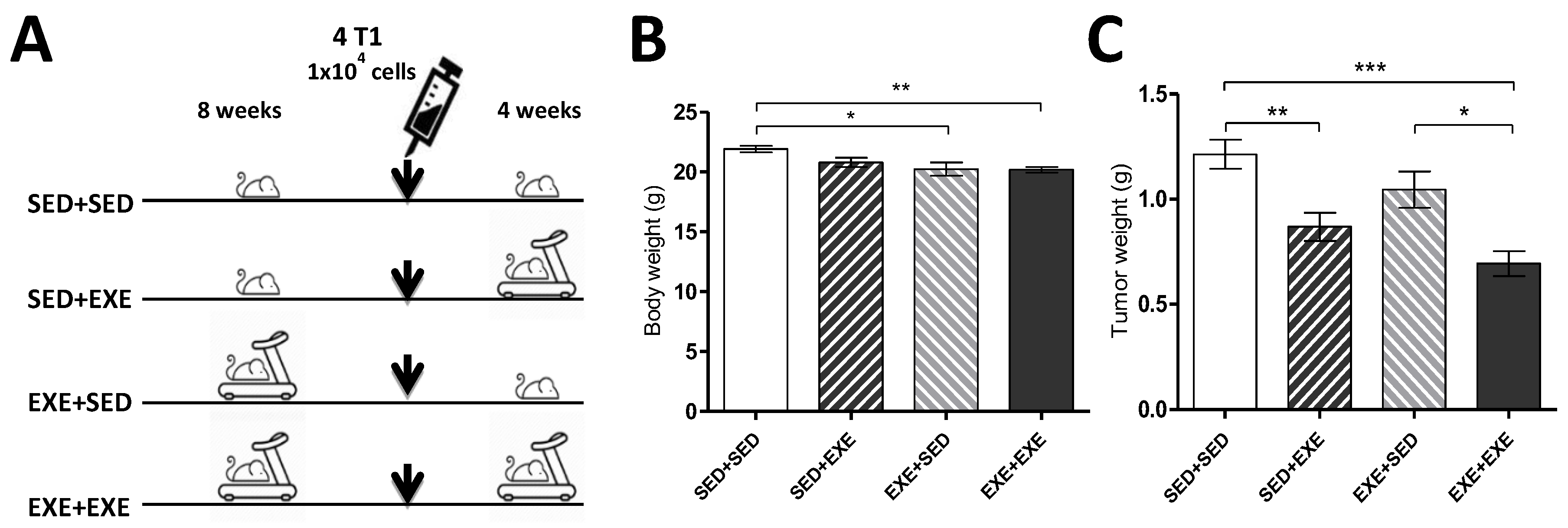
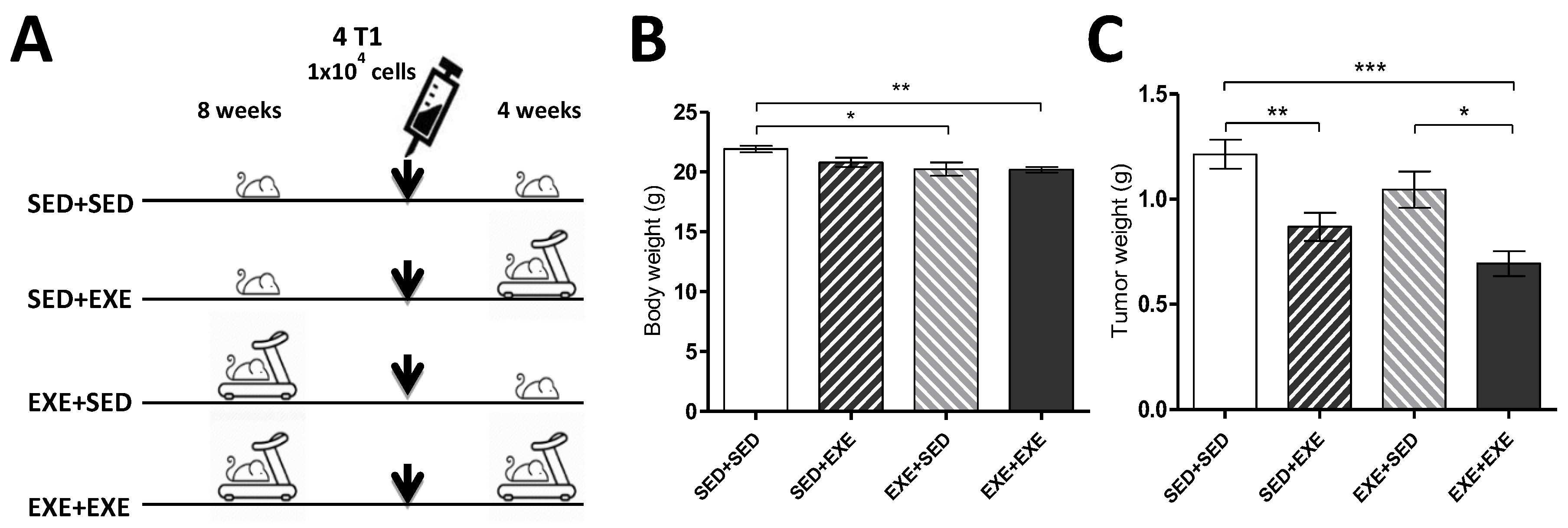
2.2. Animals and Exercise Protocols
2.3. Culture Cells
2.4. Indirect Calorimetry
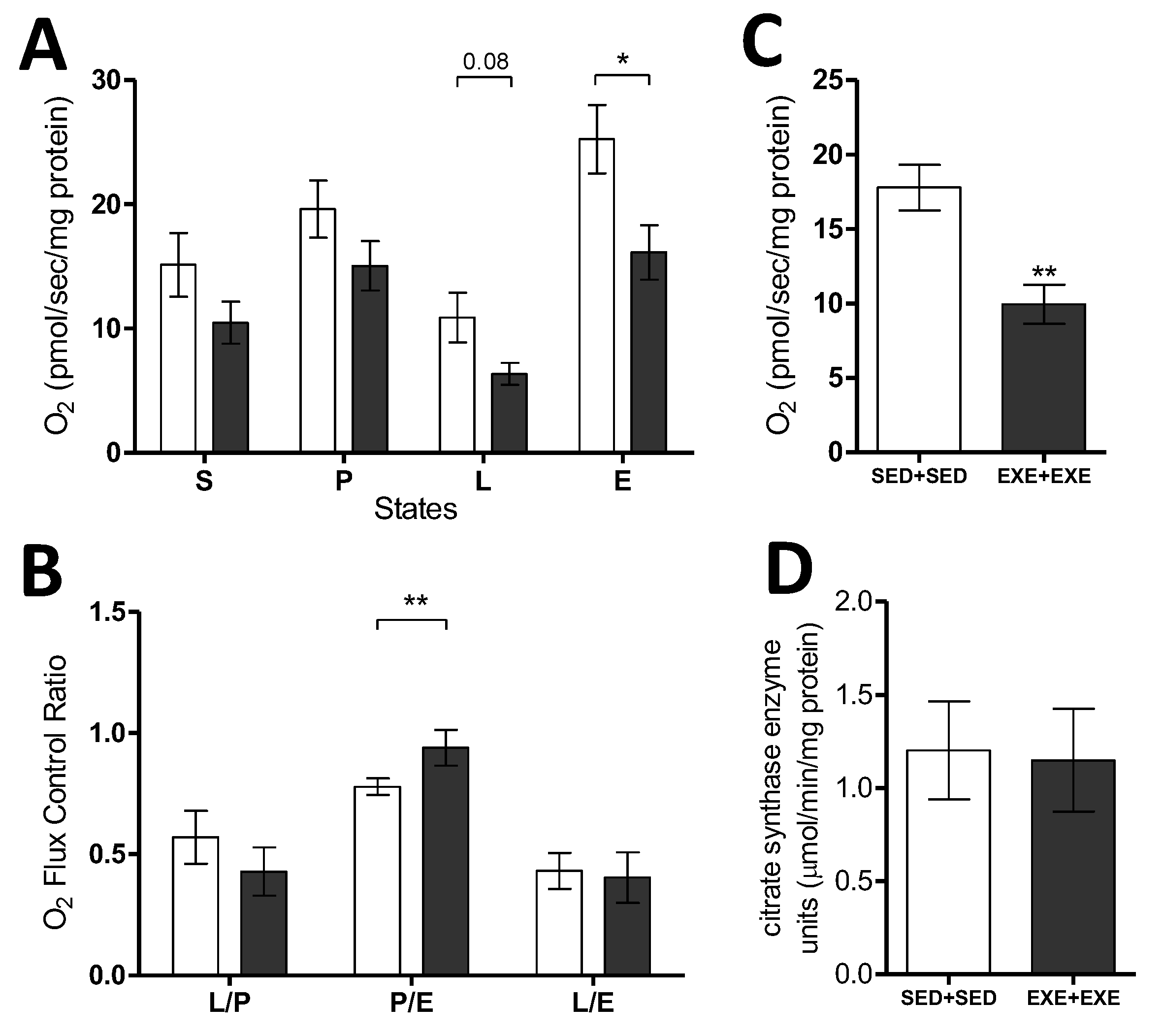
2.5. Respirometry
2.6. Citrate Synthase Activity Assay and Lactate Quantification
2.7. qPCR Analysis
2.8. Statistical Analysis
3. Results
3.1. Moderate Aerobic Exercise Reduces Body Weight and Tumor Mass
3.2. Exercise Training During the Tumorigenic Process Increases Body Carbohydrate Oxidation in the TNBC Experimental Model
3.3. Moderate Exercise Reduces Mitochondrial Respiratory Capacity in the TNBC Experimental Model
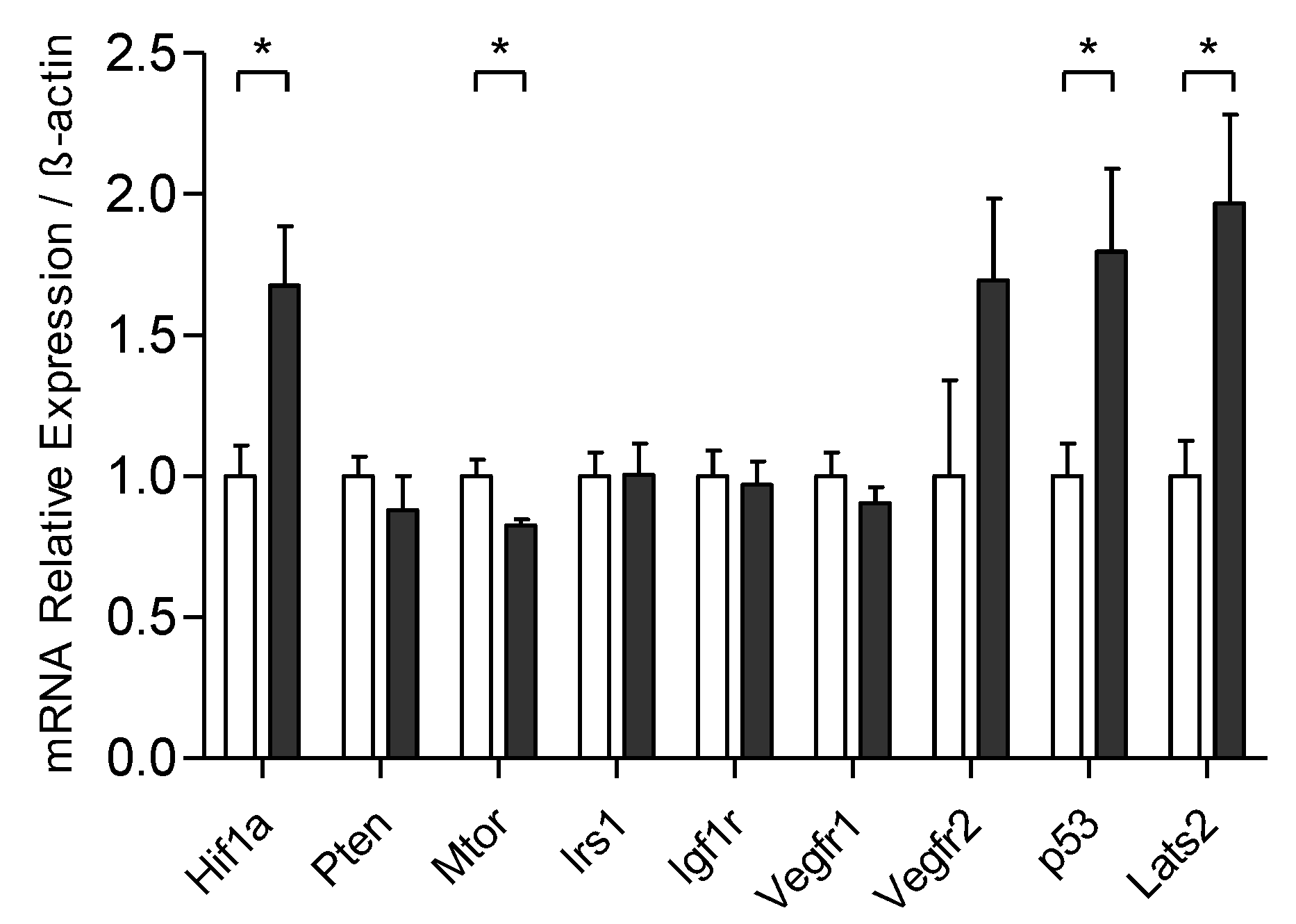
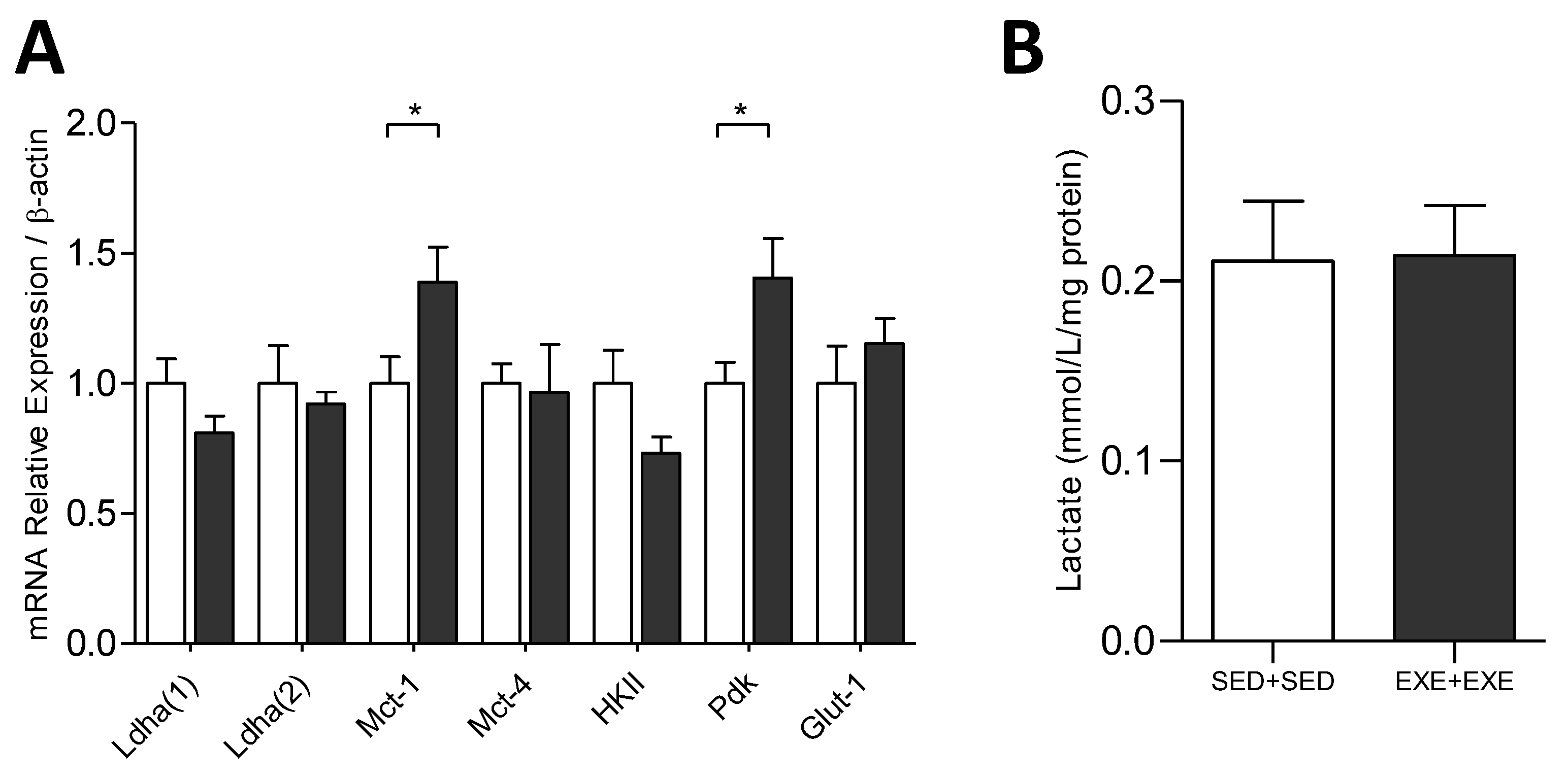
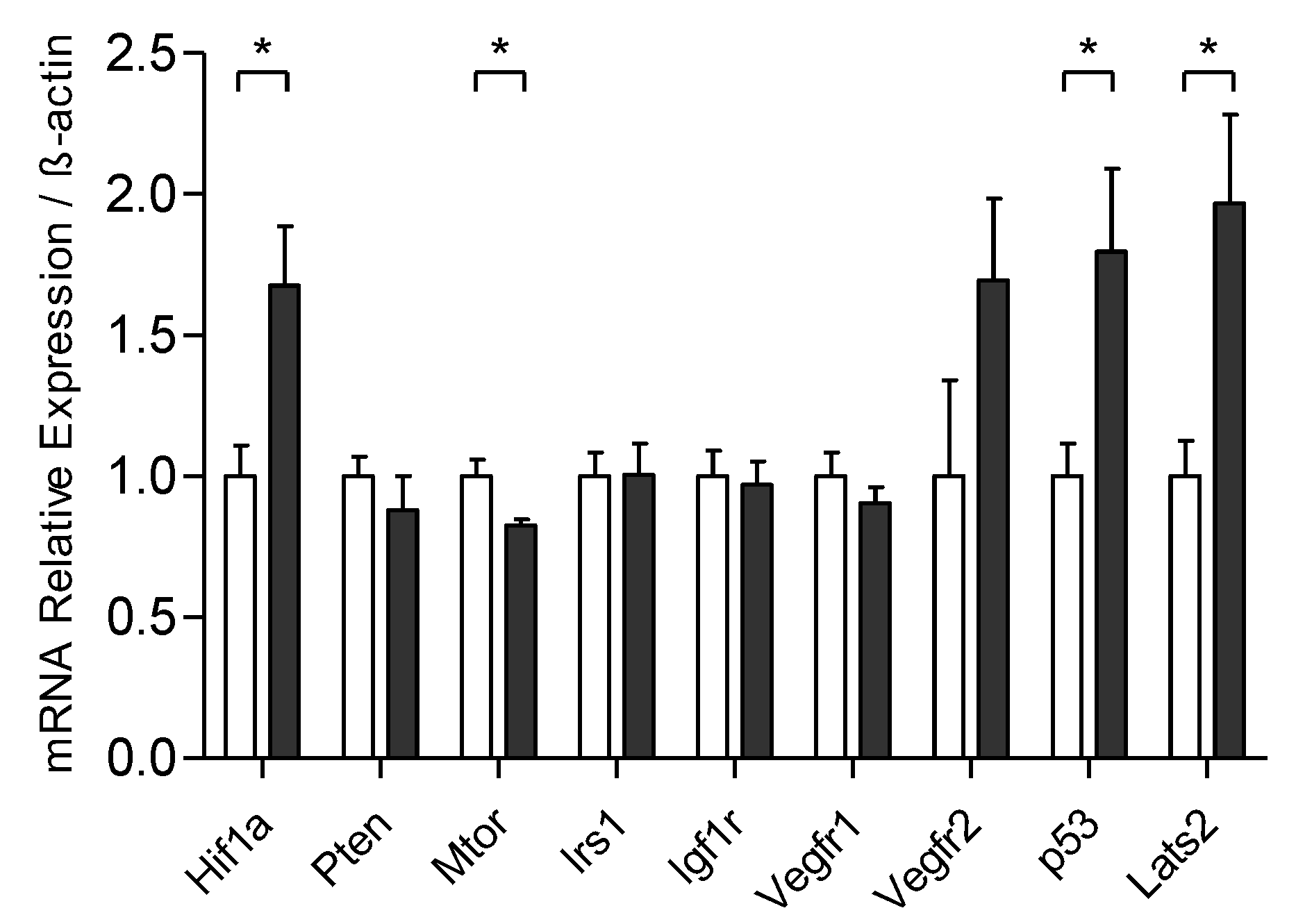
3.4. Exercise Training Modulates the mRNA Expression Related to the Control of Tumoral Process
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S.; et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, H.; He, G.; Yan, S.; Chen, C.; Song, L.; Rosol, T.J.; Deng, X. Triple-negative breast cancer: Is there a treatment on the horizon? Oncotarget 2017, 8, 1913–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aysola, K.; Desai, A.; Welch, C.; Xu, J.; Qin, Y.; Reddy, V.; Matthews, R.; Owens, C.; Okoli, J.; Beech, D.J.; et al. Triple Negative Breast Cancer—An Overview. Hered. Genet. 2013, 2013. [Google Scholar] [CrossRef]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Cancer-Genome-Atlas-Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Williams, L.A.; Butler, E.N.; Sun, X.; Allott, E.H.; Cohen, S.M.; Fuller, A.M.; Hoadley, K.A.; Perou, C.M.; Geradts, J.; Olshan, A.F.; et al. TP53 protein levels, RNA-based pathway assessment, and race among invasive breast cancer cases. NPJ Breast Cancer 2018, 4, 13. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Jose, C.; Bellance, N.; Rossignol, R. Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochim. Biophys. Acta 2011, 1807, 552–561. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. Febs J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef]
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 2018, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Lanning, N.J.; Castle, J.P.; Singh, S.J.; Leon, A.N.; Tovar, E.A.; Sanghera, A.; MacKeigan, J.P.; Filipp, F.V.; Graveel, C.R. Metabolic profiling of triple-negative breast cancer cells reveals metabolic vulnerabilities. Cancer Metab. 2017, 5, 6. [Google Scholar] [CrossRef]
- Kaur, P.; Nagaraja, G.M.; Zheng, H.; Gizachew, D.; Galukande, M.; Krishnan, S.; Asea, A. A mouse model for triple-negative breast cancer tumor-initiating cells (TNBC-TICs) exhibits similar aggressive phenotype to the human disease. BMC Cancer 2012, 12, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuy, F.; Tabaries, S.; Andrzejewski, S.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; Gao, D.; Leung, S.; Amir, E.; et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab. 2015, 22, 577–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, L.; Christensen, J.F.; Hojman, P. Effects of exercise on tumor physiology and metabolism. Cancer J. 2015, 21, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.; Idorn, M.; Olofsson, G.H.; Lauenborg, B.; Nookaew, I.; Hansen, R.H.; Johannesen, H.H.; Becker, J.C.; Pedersen, K.S.; Dethlefsen, C.; et al. Voluntary Running Suppresses Tumor Growth through Epinephrine- and IL-6-Dependent NK Cell Mobilization and Redistribution. Cell Metab. 2016, 23, 554–562. [Google Scholar] [CrossRef] [Green Version]
- Aveseh, M.; Nikooie, R.; Aminaie, M. Exercise-induced changes in tumour LDH-B and MCT1 expression are modulated by oestrogen-related receptor alpha in breast cancer-bearing BALB/c mice. J. Physiol. 2015, 593, 2635–2648. [Google Scholar] [CrossRef] [Green Version]
- Dethlefsen, C.; Pedersen, K.S.; Hojman, P. Every exercise bout matters: Linking systemic exercise responses to breast cancer control. Breast Cancer Res. Treat. 2017, 162, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Betof, A.S.; Lascola, C.D.; Weitzel, D.; Landon, C.; Scarbrough, P.M.; Devi, G.R.; Palmer, G.; Jones, L.W.; Dewhirst, M.W. Modulation of murine breast tumor vascularity, hypoxia and chemotherapeutic response by exercise. J. Natl. Cancer Inst. 2015, 107, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goh, J.; Endicott, E.; Ladiges, W.C. Pre-tumor exercise decreases breast cancer in old mice in a distance-dependent manner. Am. J. Cancer Res. 2014, 4, 378–384. [Google Scholar] [PubMed]
- Bianco, T.M.; Abdalla, D.R.; Desiderio, C.S.; Thys, S.; Simoens, C.; Bogers, J.P.; Murta, E.F.C.; Michelin, M.A. The influence of physical activity in the anti-tumor immune response in experimental breast tumor. Immunol. Lett. 2017, 190, 148–158. [Google Scholar] [CrossRef]
- Glass, O.K.; Bowie, M.; Fuller, J.; Darr, D.; Usary, J.; Boss, K.; Choudhury, K.R.; Liu, X.; Zhang, Z.; Locasale, J.W.; et al. Differential response to exercise in claudin-low breast cancer. Oncotarget 2017, 8, 100989–101004. [Google Scholar] [CrossRef] [Green Version]
- Smeda, M.; Przyborowski, K.; Proniewski, B.; Zakrzewska, A.; Kaczor, D.; Stojak, M.; Buczek, E.; Nieckarz, Z.; Zoladz, J.A.; Wietrzyk, J.; et al. Breast cancer pulmonary metastasis is increased in mice undertaking spontaneous physical training in the running wheel; a call for revising beneficial effects of exercise on cancer progression. Am. J. Cancer Res. 2017, 7, 1926–1936. [Google Scholar]
- Pesta, D.; Gnaiger, E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol 2012, 810, 25–58. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Schneeberger, S.; Seiler, R.; Brandacher, G.; Mark, W.; Steurer, W.; Saks, V.; Usson, Y.; Margreiter, R.; Gnaiger, E. Mitochondrial defects and heterogeneous cytochrome c release after cardiac cold ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1633–H1641. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, D.; Garland, P.B. ATP controlled acetoacetate and citrate synthesis by rat liver mitochondria oxidising palmitoyl-carnitine, and the inhibition of citrate synthase by ATP. Biochem. Biophys. Res. Commun. 1966, 22, 89–93. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Haugen, H.A.; Chan, L.N.; Li, F. Indirect calorimetry: A practical guide for clinicians. Nutr. Clin. Pr. 2007, 22, 377–388. [Google Scholar] [CrossRef]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371; e359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molanouri Shamsi, M.; Chekachak, S.; Soudi, S.; Quinn, L.S.; Ranjbar, K.; Chenari, J.; Yazdi, M.H.; Mahdavi, M. Combined effect of aerobic interval training and selenium nanoparticles on expression of IL-15 and IL-10/TNF-α ratio in skeletal muscle of 4T1 breast cancer mice with cachexia. Cytokine 2017, 90, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Lu, M.; Jia, D.; Ma, J.; Ben-Jacob, E.; Levine, H.; Kaipparettu, B.A.; Onuchic, J.N. Modeling the Genetic Regulation of Cancer Metabolism: Interplay between Glycolysis and Oxidative Phosphorylation. Cancer Res. 2017, 77, 1564–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife 2017, 6, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Graham, K.A.; Kulawiec, M.; Owens, K.M.; Li, X.; Desouki, M.M.; Chandra, D.; Singh, K.K. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol. 2010, 10, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Panagopoulos, V.; Leach, D.A.; Zinonos, I.; Ponomarev, V.; Licari, G.; Liapis, V.; Ingman, W.V.; Anderson, P.; DeNichilo, M.O.; Evdokiou, A. Inflammatory peroxidases promote breast cancer progression in mice via regulation of the tumour microenvironment. Int. J. Oncol. 2017, 50, 1191–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, L.W.; Viglianti, B.L.; Tashjian, J.A.; Kothadia, S.M.; Keir, S.T.; Freedland, S.J.; Potter, M.Q.; Moon, E.J.; Schroeder, T.; Herndon, J.E., 2nd; et al. Effect of aerobic exercise on tumor physiology in an animal model of human breast cancer. J. Appl. Physiol. (1985) 2010, 108, 343–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isanejad, A.; Alizadeh, A.M.; Amani Shalamzari, S.; Khodayari, H.; Khodayari, S.; Khori, V.; Khojastehnjad, N. MicroRNA-206, let-7a and microRNA-21 pathways involved in the anti-angiogenesis effects of the interval exercise training and hormone therapy in breast cancer. Life Sci. 2016, 151, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, C.; Hansen, L.S.; Lillelund, C.; Andersen, C.; Gehl, J.; Christensen, J.F.; Pedersen, B.K.; Hojman, P. Exercise-Induced Catecholamines Activate the Hippo Tumor Suppressor Pathway to Reduce Risks of Breast Cancer Development. Cancer Res. 2017, 77, 4894–4904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Zhou, Z.; Flesken-Nikitin, A.; Toshkov, I.A.; Wang, W.; Camps, J.; Ried, T.; Nikitin, A.Y. Rb inactivation accelerates neoplastic growth and substitutes for recurrent amplification of cIAP1, cIAP2 and Yap1 in sporadic mammary carcinoma associated with p53 deficiency. Oncogene 2010, 29, 5700–5711. [Google Scholar] [CrossRef] [Green Version]
- Furth, N.; Aylon, Y.; Oren, M. p53 shades of Hippo. Cell Death Differ. 2018, 25, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Ardestani, A.; Lupse, B.; Maedler, K. Hippo Signaling: Key Emerging Pathway in Cellular and Whole-Body Metabolism. Trends Endocrinol. Metab. 2018, 29, 492–509. [Google Scholar] [CrossRef]
- Jamurtas, A.Z.; Koutedakis, Y.; Paschalis, V.; Tofas, T.; Yfanti, C.; Tsiokanos, A.; Koukoulis, G.; Kouretas, D.; Loupos, D. The Effects of a Single Bout of Exercise on Resting Energy Expenditure and Respiratory Exchange Ratio. Eur. J. Appl. Physiol. 2004, 92. [Google Scholar] [CrossRef] [Green Version]
- Nakagata, T.; Yamada, Y.; Naito, H. Energy Expenditure, Recovery Oxygen Consumption, and Substrate Oxidation During and After Body Weight Resistance Exercise With Slow Movement Compared to Treadmill Walking. Physiol. Int. 2018, 105. [Google Scholar] [CrossRef]
- Lu, M.; Sanderson, S.M.; Zessin, A.; Ashcraft, K.A.; Jones, L.W.; Dewhirst, M.W.; Locasale, J.W.; Hsu, D.S. Exercise inhibits tumor growth and central carbon metabolism in patient-derived xenograft models of colorectal cancer. Cancer Metab. 2018, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Serganova, I.; Cohen, I.J.; Vemuri, K.; Shindo, M.; Maeda, M.; Mane, M.; Moroz, E.; Khanin, R.; Satagopan, J.; Koutcher, J.A.; et al. LDH-A regulates the tumor microenvironment via HIF-signaling and modulates the immune response. PLoS ONE 2018, 13, e0203965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, H.; Hanai, J.; Ren, J.G.; Kats, L.; Burgess, K.; Bhargava, P.; Signoretti, S.; Billiard, J.; Duffy, K.J.; Grant, A.; et al. Targeting lactate dehydrogenase—A inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 2014, 19, 795–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| Ldha iso1 | ATCGTGCACTAGCGGTCTCA | CCATCATCTCGCCCTTGAGT |
| Ldha iso2 | AACTTGGCGCTCTACTTGCT | GGACTTTGAATCTTTTGAGACCTTG |
| Mct1 | ACGCCGGAGTCTTTGGATTT | TGAGGCGGCCTAAAAGTGG |
| Mct4 | GCACTTAAAGTCGCCCCCG | GAGGGCTGCTTTCACCAAGAAC |
| HKII | GGAGTCTTCGATCCCAGCCG | CTGGTCAACCTTCTGCACTTGG |
| Pdk 1 | AAGCAGTTCCTGGACTTCGG | GGCTTTGGATATACCAACTTTGC |
| Glut-1 | GTGACGATCTGAGCTACGGG | TCACCTTCTTGCTGCTGGG |
| Hif1a | TGAGTTCTGAACGTCGAAAAGA | TAGACCACCGGCATCCAGA |
| Pten | CAGGCTCCCAGACATGACA | GCTTTGAATCCAAAAACCTTACTAC |
| Mtor | TCACTTCCTGAACAGCGAGC | GTAGCGGATATCAGGGTCAGG |
| Irs-1 | CCGATATGGTGATGAGGAGCTG | TGGCAATATTTGATGGGACATCT |
| Igf1r | GCACCAATGCTTCAGTCCCT | TTGGAGCAGTAGTTGTGCCG |
| Vegfr 1 | GTGTCTATAGGTGCCGAGCC | CGGAAGAAGACCGCTTCAGT |
| Vegfr 2 | GCATACCGCCTCTGTGACTT | AAATCGCCAGGCAAACCCAC |
| p53 | TCCGAAGACTGGATGACTGC | GCTTCACTTGGGCCTTCAAA |
| Lats 2 | AGCTGAAGGTGATCAACTGGG | CAGGCTGCTTTCGGATGTCA |
| β Actina | GATCAAGATCATTGCTCCTCCTG | AGGGTGTAAAACGCAGCTCA |
| (SED + SED) | (SED + EXE) | (EXE + SED) | (EXE + EXE) | |
|---|---|---|---|---|
| RER pretumor | 0.98 ± 0.08 | 0.97 ± 0.11 | 0.90 ± 0.11 a,b | 0.89 ± 0.10 a,b |
| RER post-tumor | 0.92 ± 0.12† | 1.00 ± 0.07 a,d,‡ | 0.88 ± 0.14 b,c | 0.98 ± 0.09 a,d,‡ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vulczak, A.; Souza, A.d.O.; Ferrari, G.D.; Azzolini, A.E.C.S.; Pereira-da-Silva, G.; Alberici, L.C. Moderate Exercise Modulates Tumor Metabolism of Triple-Negative Breast Cancer. Cells 2020, 9, 628. https://doi.org/10.3390/cells9030628
Vulczak A, Souza AdO, Ferrari GD, Azzolini AECS, Pereira-da-Silva G, Alberici LC. Moderate Exercise Modulates Tumor Metabolism of Triple-Negative Breast Cancer. Cells. 2020; 9(3):628. https://doi.org/10.3390/cells9030628
Chicago/Turabian StyleVulczak, Anderson, Anderson de Oliveira Souza, Gustavo Duarte Ferrari, Ana Elisa Caleiro Seixas Azzolini, Gabriela Pereira-da-Silva, and Luciane Carla Alberici. 2020. "Moderate Exercise Modulates Tumor Metabolism of Triple-Negative Breast Cancer" Cells 9, no. 3: 628. https://doi.org/10.3390/cells9030628