A STAT3 of Addiction: Adipose Tissue, Adipocytokine Signalling and STAT3 as Mediators of Metabolic Remodelling in the Tumour Microenvironment


Abstract
:1. Introduction
2. Cancer: An Interplay of Canonical and Non-Canonical STAT3 Signalling
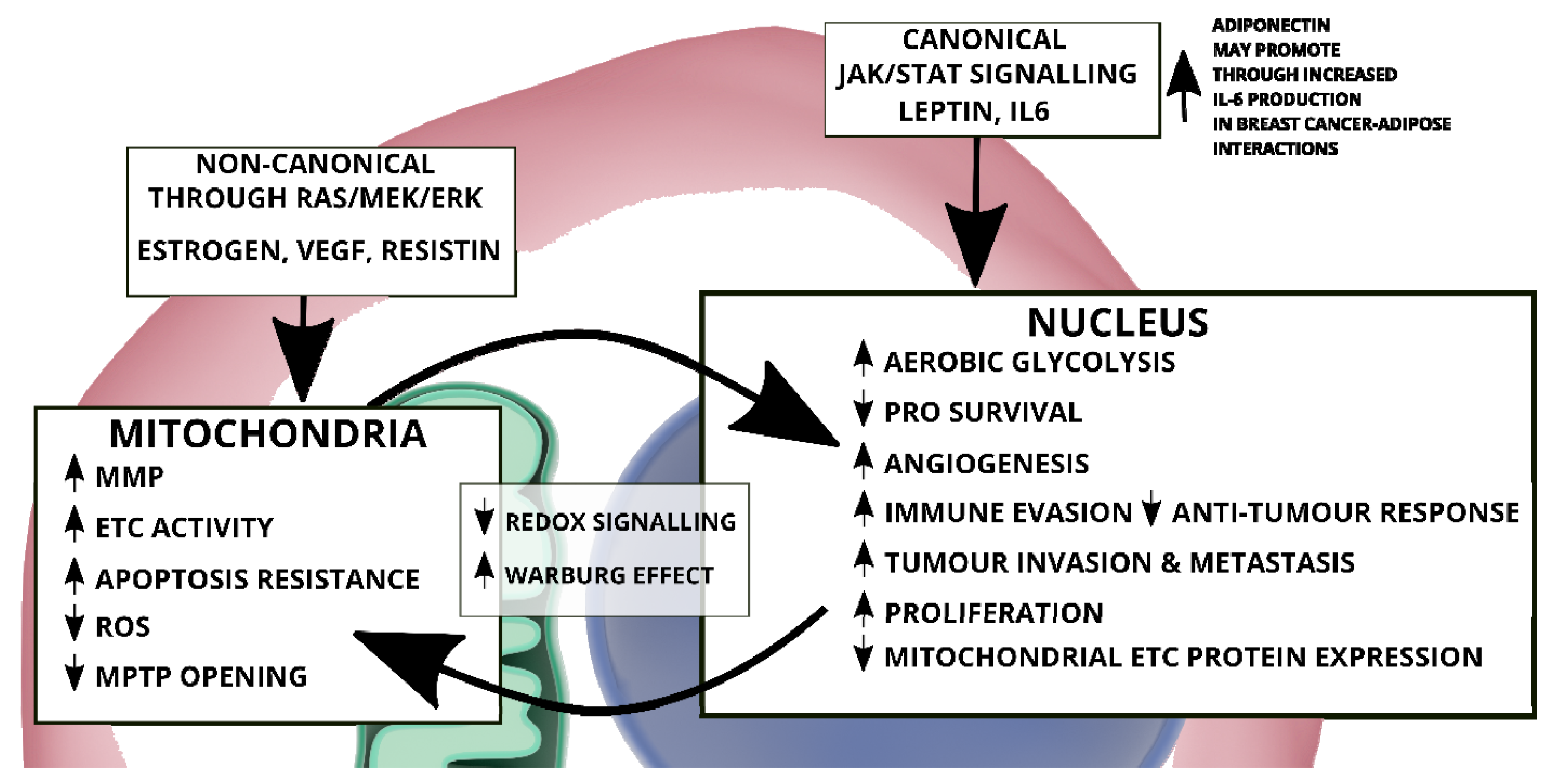
2.1. Canonical STAT3 Signalling and the Warburg Effect
2.2. Mitochondrial STAT3: Inside the Engine Core
2.3. Metabolic Remodelling through STAT3 Stimulation
3. REDOX Signalling in Cancer: Does STAT3 Maintain the Balance?
4. STAT3 as a Regulatory Buffer of Apoptosis and Autophagy
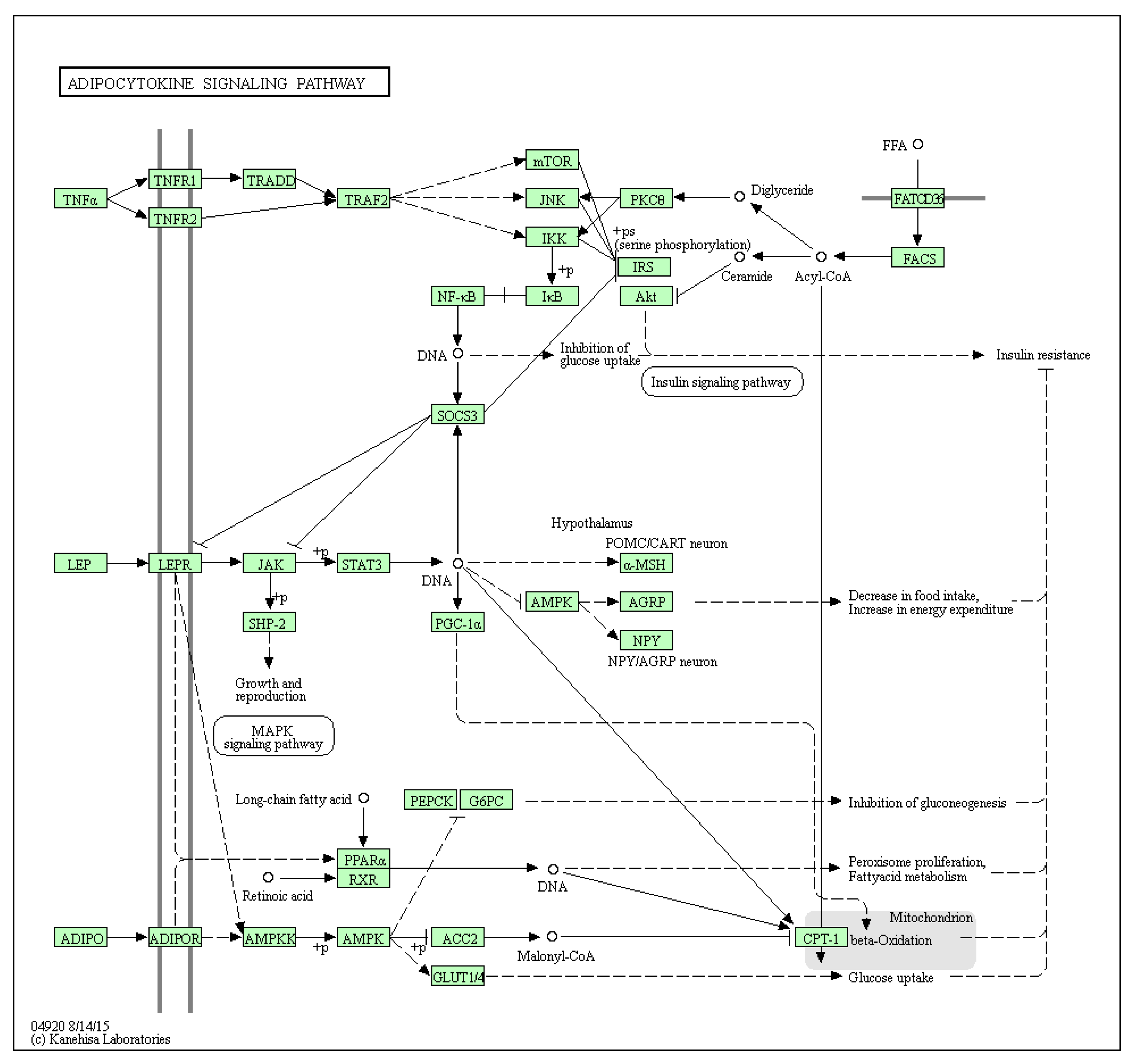
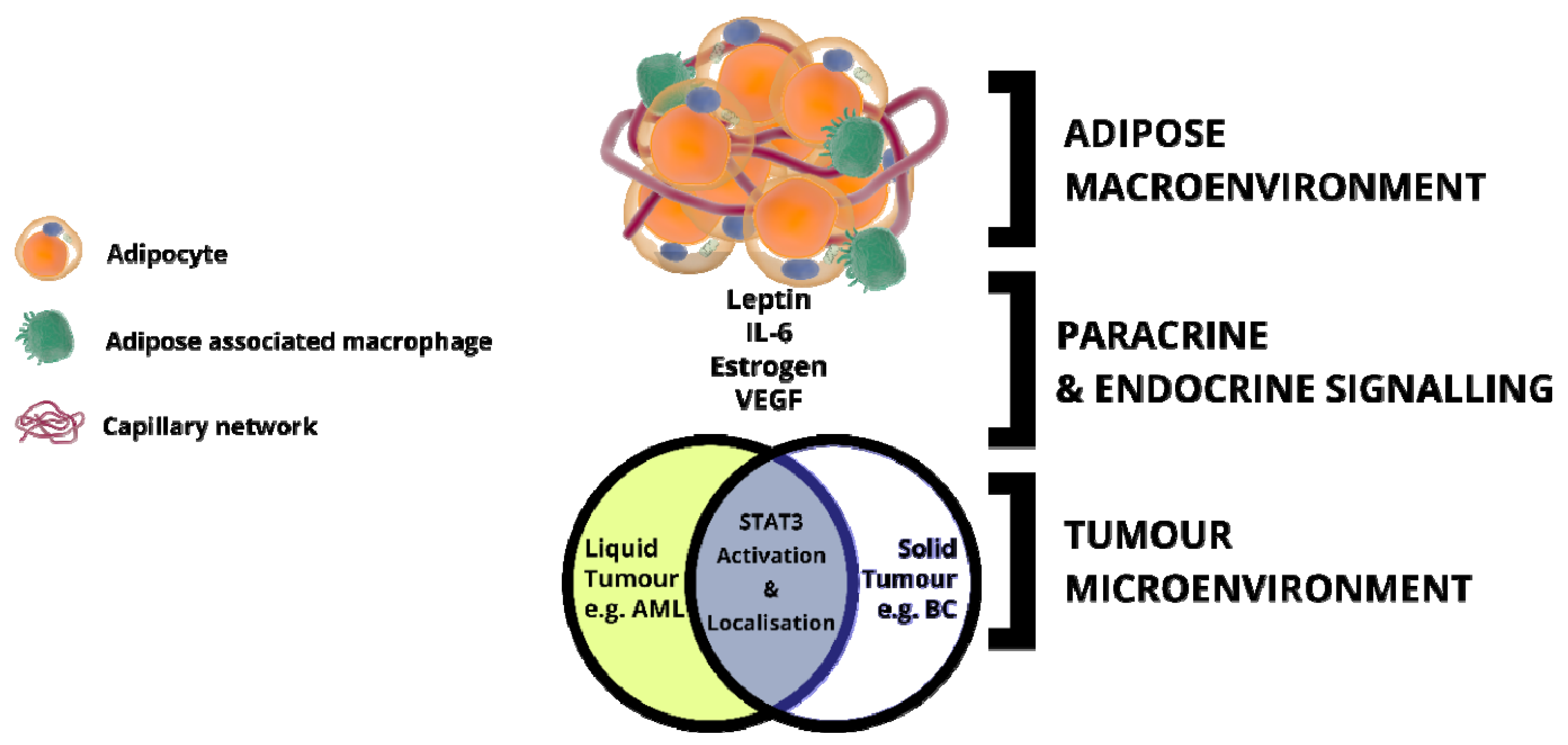
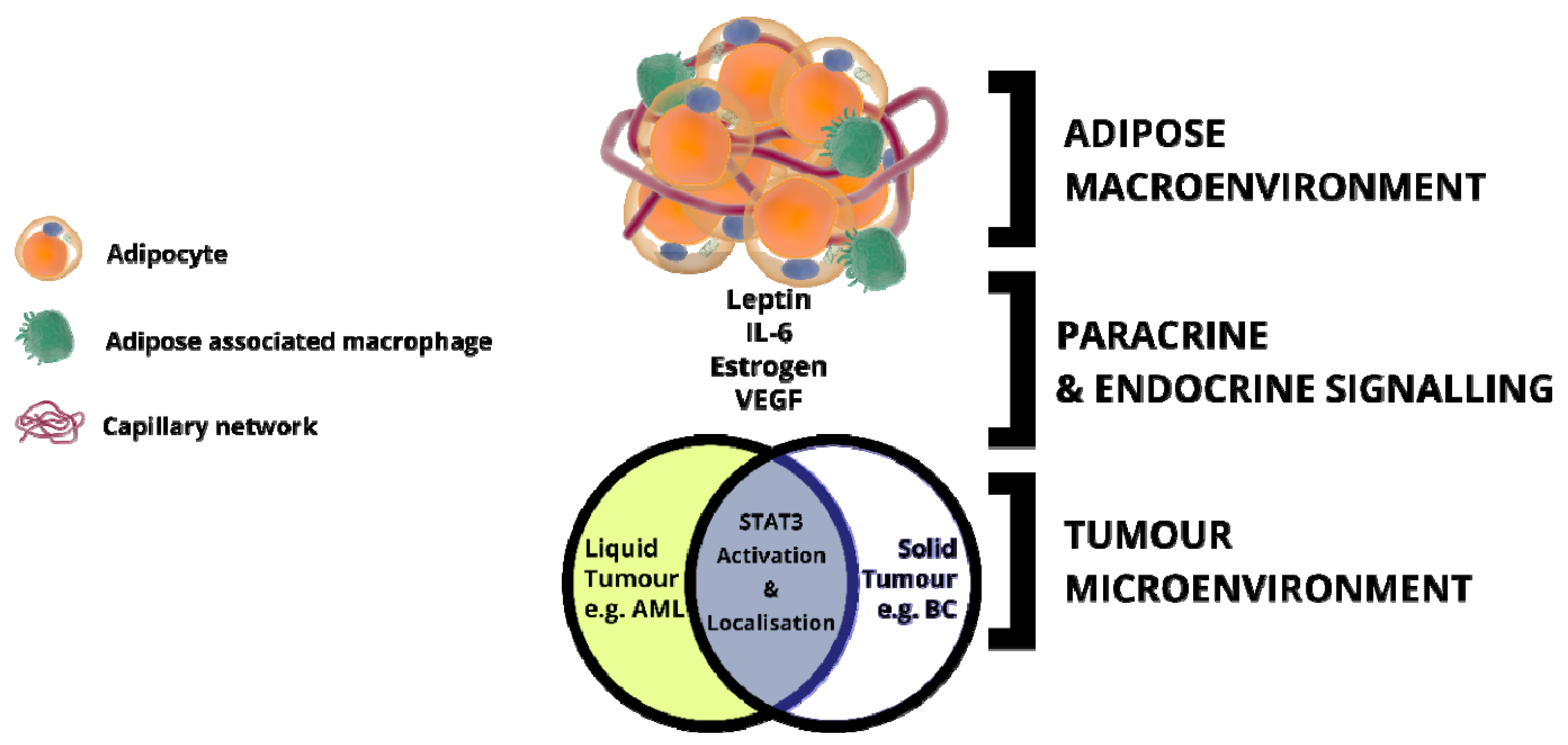
5. Adipose Tissue: An Inflammatory Macroenvironment Stimulating the Tumour Microenvironment through STAT3
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wegrzyn, J.; Potla, R.; Chwae, Y.Y.-J.; Sepuri, N.N.B.V.V.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar] [PubMed]
- Himbert, C.; Delphan, M.; Scherer, D.; Bowers, L.W.; Hursting, S.; Ulrich, C.M. Signals from the adipose microenvironment and the obesity-cancer link-a systematic review. Cancer Prev. Res. 2017, 10, 494–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duong, M.N.; Geneste, A.; Fallone, F.; Li, X.; Dumontet, C.; Muller, C. The fat and the bad: Mature adipocytes, key actors in tumor progression and resistance. Oncotarget 2017, 8, 57622–57641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoico, E.; Darra, E.; Rizzatti, V.; Tebon, M.; Franceschetti, G.; Mazzali, G.; Rossi, A.P.; Fantin, F.; Zamboni, M. Role of adipose tissue in melanoma cancer microenvironment and progression. Int. J. Obes. 2018, 42, 344–352. [Google Scholar] [CrossRef]
- Huang, S. Regulation of metastases by signal transducer and activator of transcription 3 signaling pathway: Clinical implications. Clin. Cancer Res. 2007, 13, 1362–1366. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Kamran, M.Z.; Patil, P.; Gude, R.P. Role of STAT3 in cancer metastasis and translational advances. Biomed. Res. Int. 2013, 2013, 421821. [Google Scholar] [CrossRef]
- Aggarwal, B.; Heber, D. Immunonutrition. Interactions of Diet, Genetics, and Inflammation, 1st ed.; CRC Press: Boca Raton, FL, USA, 2014; pp. 1–38. [Google Scholar]
- Shalapour, S.; Karin, M. Immunity, inflammation, and cancer: An eternal fight between good and evil. J. Clin. Investig. 2015, 125, 3347–3355. [Google Scholar] [CrossRef] [Green Version]
- Kamp, D.W.; Shacter, E.; Weitzman, S.A. Chronic inflammation and cancer: The role of the mitochondria. Oncology 2011, 25, 400–410, 413. [Google Scholar] [PubMed]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timofeeva, O.A.; Chasovskikh, S.; Lonskaya, I.; Tarasova, N.I.; Khavrutskii, L.; Tarasov, S.G.; Zhang, X.; Korostyshevskiy, V.R.; Cheema, A.; Zhang, L.; et al. Mechanisms of Unphosphorylated STAT3 Transcription Factor Binding to DNA. J. Biol. Chem. 2012, 287, 14192–14200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFκB. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hnisz, D.; Schuijers, J.; Lin, C.Y.; Weintraub, A.S.; Abraham, B.J.; Lee, T.I.; Bradner, J.E.; Young, R.A. Convergence of Developmental and Oncogenic Signaling Pathways at Transcriptional Super-Enhancers. Mol. Cell 2015, 58, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Finger, E.C.; Giaccia, A.J. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev. 2010, 29, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.Y.; Liu, S.C.; Johnson, D.G.; Klein-Szanto, A.J.P. E2F-1 gene transfer enhances invasiveness of human head and neck carcinoma cell lines. Cancer Res. 2000, 60, 5972–5976. [Google Scholar]
- Cameron, I.L.; Markov, M.S.; Hardman, W.E. Optimization of a therapeutic electromagnetic field (EMF) to retard breast cancer tumor growth and vascularity. Cancer Cell Int. 2014, 14, 125. [Google Scholar] [CrossRef] [Green Version]
- Muz, B.; De la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, Y.; Shingu, T.; Feng, L.; Chen, Z.; Ogasawara, M.; Keating, M.J.; Kondo, S.; Huang, P. Metabolic alterations in highly tumorigenic glioblastoma cells: Preference for hypoxia and high dependency on glycolysis. J. Biol. Chem. 2011, 286, 32843–32853. [Google Scholar] [CrossRef] [Green Version]
- Corbet, C.; Feron, O. Cancer cell metabolism and mitochondria: Nutrient plasticity for TCA cycle fueling. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria in cancer cells: What is so special about them? Trends Cell Biol. 2008, 18, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.H.; Chen, S.P.; Mao, J.Y.; Ji, X.X.; Yao, H.T.; Zhou, S.H. Expression and significance of hypoxia-inducible factor-1α and glucose transporter-1 in laryngeal carcinoma. Oncol. Lett. 2012, 8, 57622–57641. [Google Scholar] [CrossRef] [Green Version]
- Luis, C.; Duarte, F.; Faria, I.; Jarak, I.; Oliveira, P.F.; Alves, M.G.; Soares, R.; Fernandes, R. Warburg Effect Inversion: Adiposity shifts central primary metabolism in MCF-7 breast cancer cells. Life Sci. 2019, 223, 38–46. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.P.; Sabatini, D.M. Cancer Cell Metabolism: Warburg and Beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.-Y.; Ann, D.K. When fats commit crimes: Fatty acid metabolism, cancer stemness and therapeutic resistance. Cancer Commun. 2018, 38, 47. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-T.; Hsu, S.-H.; Wei, Y.-H. Mitochondrial bioenergetic function and metabolic plasticity in stem cell differentiation and cellular reprogramming. Biochim. Biophys. Acta 2012, 1820, 571–576. [Google Scholar] [CrossRef]
- Darnell, J.; Kerr, I.; Stark, G. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, D.E.; Lee, C.-K. What does Stat3 do? J. Clin. Invest. 2002, 109, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, P.B.; Kumar, V.; Guo, G.; Murray, W.C. Different patterns of regulation of Tyr-phosphorylated STAT1 and STAT3 in human hepatoma Hep3B cells by the phosphatase inhibitor orthovanadate. Arch. Biochem. Biophys. 2003, 412, 242–250. [Google Scholar] [CrossRef]
- Xu, F.; Mukhopadhyay, S.; Sehgal, P.B. Live cell imaging of interleukin-6-induced targeting of “transcription factor” STAT3 to sequestering endosomes in the cytoplasm. Am. J. Physiol. Cell Physiol. 2007, 293, C1374–C1382. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. STAT family of transcription factors in cytokine-mediated biological responses. Cytokine Growth Factor Rev. 2000, 11, 199–207. [Google Scholar] [CrossRef]
- Aaronson, D.S.; Horvath, C.M. The STAT3 Pathway. Sci. Signal 2003, 2003, cm13. [Google Scholar] [CrossRef]
- Yuan, J.; Yang, Y.; Gao, Z.; Wang, Z.; Ji, W.; Song, W.; Zhang, F.; Niu, R. Tyr23 phosphorylation of Anxa2 enhances STAT3 activation and promotes proliferation and invasion of breast cancer cells. Breast Cancer Res. Treat. 2017, 164, 327–340. [Google Scholar] [CrossRef]
- Khan, M.W.; Saadalla, A.; Ewida, A.H.; Al-Katranji, K.; Al-Saoudi, G.; Giaccone, Z.T.; Gounari, F.; Zhang, M.; Frank, D.A.; Khazaie, K. The STAT3 inhibitor pyrimethamine displays anti-cancer and immune stimulatory effects in murine models of breast cancer. Cancer Immunol. Immunother. 2018, 67, 13–23. [Google Scholar] [CrossRef]
- McDaniel, J.M.; Varley, K.E.; Gertz, J.; Savic, D.S.; Roberts, B.S.; Bailey, S.K.; Shevde, L.A.; Ramaker, R.C.; Lasseigne, B.N.; Kirby, M.K.; et al. Genomic regulation of invasion by STAT3 in triple negative breast cancer. Oncotarget 2017, 8, 8226–8238. [Google Scholar] [CrossRef] [Green Version]
- Oh, E.; Kim, Y.J.; An, H.; Sung, D.; Cho, T.M.; Farrand, L.; Jang, S.; Seo, J.H.; Kim, J.Y. Flubendazole elicits anti-metastatic effects in triple-negative breast cancer via STAT3 inhibition. Int. J. Cancer 2018, 143, 1978–1993. [Google Scholar] [CrossRef] [Green Version]
- Balanis, N.; Carlin, C.R. Stress-induced EGF receptor signaling through STAT3 and tumor progression in triple-negative breast cancer. Mol. Cell Endocrinol. 2017, 451, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.E.; Lee, H.G.; Cho, I.H.; Chung, D.H.; Yoon, S.-H.; Yang, Y.M.; Lee, J.W.; Choi, S.; Park, J.-W.; Ye, S.-K.; et al. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J. 2005, 19, 1296–1298. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E. STAT3, HIF-1, glucose addiction and Warburg effect. Aging 2010, 2, 890–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egusquiaguirre, S.P.; Yeh, J.E.; Walker, S.R.; Liu, S.; Frank, D.A. The STAT3 Target Gene TNFRSF1A Modulates the NF-κB Pathway in Breast Cancer Cells. Neoplasia 2018, 20, 489–498. [Google Scholar] [CrossRef]
- Dreesen, O.; Brivanlou, A.H. Signaling Pathways in Cancer and Embryonic Stem Cells. Stem Cell Rev. 2007, 3, 7–17. [Google Scholar] [CrossRef]
- Bromberg, J.; Wrzeszczynska, M.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E. Stat3 as an Oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Demaria, M.; Poli, V. PKM2, STAT3 and HIF-1α. JAKSTAT 2012, 1, 194–196. [Google Scholar] [CrossRef] [Green Version]
- Demaria, M.; Giorgi, C.; Lebiedzinska, M.; Esposito, G.; D’Angeli, L.; Bartoli, A.; Gough, D.J.; Turkson, J.; Levy, D.E.; Watson, C.J.; et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging 2010, 2, 823–842. [Google Scholar] [CrossRef] [Green Version]
- Aghazadeh, S.; Yazdanparast, R. Activation of STAT3/HIF-1α/Hes-1 axis promotes trastuzumab resistance in HER2-overexpressing breast cancer cells via down-regulation of PTEN. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1970–1980. [Google Scholar] [CrossRef]
- Wincewicz, A.; Koda, M.; Sulkowska, M.; Kanczuga-Koda, L.; Wincewicz, D.; Sulkowski, S. STAT3 and hypoxia induced proteins--HIF-1alpha, EPO and EPOR in relation with Bax and Bcl-xL in nodal metastases of ductal breast cancers. Folia Histochem. Cytobiol. 2010, 47, 425–430. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, F.; Niu, R. Multiple regulation pathways and pivotal biological functions of STAT3 in cancer. Sci. Rep. 2015, 5, 17663. [Google Scholar] [CrossRef] [Green Version]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Jiang, H.; Xu, X.; Duan, W.; Mattson, M.P. Leptin-mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. J. Biol. Chem. 2008, 283, 1754–1763. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, B.B.; Kunnumakkara, A.B.; Harikumar, K.B.; Gupta, S.R.; Tharakan, S.T.; Koca, C.; Dey, S.; Sung, B. Signal transducer and activator of transcription-3, inflammation, and cancer: How intimate is the relationship? Ann. NY Acad. Sci. 2009, 1171, 59–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Wang, L.H.; Farrar, W.L. Interleukin 6 activates androgen receptor-mediated gene expression through a signal transducer and activator of transcription 3-dependent pathway in LNCaP prostate cancer cells. Cancer Res. 2000, 60, 2132–2135. [Google Scholar] [PubMed]
- Avalle, L.; Camporeale, A.; Camperi, A.; Poli, V. STAT3 in cancer: A double edged sword. Cytokine 2017, 98, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 Activation by IL-6 Transsignaling Promotes Progression of Pancreatic Intraepithelial Neoplasia and Development of Pancreatic Cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef] [Green Version]
- Niemand, C.; Nimmesgern, A.; Haan, S.; Fischer, P.; Schaper, F.; Rossaint, R.; Heinrich, P.C.; Müller-Newen, G. Activation of STAT3 by IL-6 and IL-10 in Primary Human Macrophages Is Differentially Modulated by Suppressor of Cytokine Signaling 3. J. Immunol. 2003, 170, 3263–3272. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Uddin, N.; Gribben, J.G. Activation of Mitochondrial STAT3 Increases Mitochondrial Respiration and Inhibits Oxidative Stress in Chronic Lymphocytic Leukemic Cells. Blood 2011, 118, 287. [Google Scholar] [CrossRef]
- Velichko, S.; Wagner, T.C.; Turkson, J.; Jove, R.; Croze, E. STAT3 Activation by Type I Interferons Is Dependent on Specific Tyrosines Located in the Cytoplasmic Domain of Interferon Receptor Chain 2c. J. Biol. Chem. 2002, 277, 35635–35641. [Google Scholar] [CrossRef] [Green Version]
- Nguyen-Jackson, H.T.; Li, H.S.; Zhang, H.; Ohashi, E.; Watowich, S.S. G-CSF-activated STAT3 enhances production of the chemokine MIP-2 in bone marrow neutrophils. J. Leukoc. Biol. 2012, 92, 1215–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiki, S.; Hato, F.; Kamata, N.; Sakamoto, E.; Hasegawa, T.; Kimura-Eto, A.; Hino, M.; Kitagawa, S. Selective activation of STAT3 in human monocytes stimulated by G-CSF: Implication in inhibition of LPS-induced TNF-α production. Am. J. Physiol. Cell Physiol. 2004, 286, C1302–C1311. [Google Scholar] [CrossRef] [PubMed]
- Park, O.K.; Schaefer, T.S.; Nathans, D. In vitro activation of Stat3 by epidermal growth factor receptor kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 13704–13708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-Z.; Wharton, W.; Garcia, R.; Kraker, A.; Jove, R.; Pledger, W.J. Activation of Stat3 preassembled with platelet-derived growth factor β receptors requires Src kinase activity. Oncogene 2000, 19, 2075–2085. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Wen, Z.; Darnell, J.E. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef]
- Vignais, M.L.; Sadowski, H.B.; Watling, D.; Rogers, N.C.; Gilman, M. Platelet-derived growth factor induces phosphorylation of multiple JAK family kinases and STAT proteins. Mol. Cell. Biol. 1996, 16, 1759–1769. [Google Scholar] [CrossRef] [Green Version]
- Guryanova, O.A.; Wu, Q.; Cheng, L.; Lathia, J.D.; Huang, Z.; Yang, J.; MacSwords, J.; Eyler, C.E.; McLendon, R.E.; Heddleston, J.M.; et al. Nonreceptor Tyrosine Kinase BMX Maintains Self-Renewal and Tumorigenic Potential of Glioblastoma Stem Cells by Activating STAT3. Cancer Cell 2011, 19, 498–511. [Google Scholar] [CrossRef] [Green Version]
- Garcia, R.; Yu, C.L.; Hudnall, A.; Catlett, R.; Nelson, K.L.; Smithgall, T.; Fujita, D.J.; Ethier, S.P.; Jove, R. Constitutive activation of Stat3 in fibroblasts transformed by diverse oncoproteins and in breast carcinoma cells. Cell Growth Differ. 1997, 8, 1267–1276. [Google Scholar]
- Zhang, X.; Guo, A.; Yu, J.; Possemato, A.; Chen, Y.; Zheng, W.; Polakiewicz, R.D.; Kinzler, K.W.; Vogelstein, B.; Velculescu, V.E.; et al. Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc. Natl. Acad. Sci. USA 2007, 104, 4060–4064. [Google Scholar] [CrossRef] [Green Version]
- Veeriah, S.; Brennan, C.; Meng, S.; Singh, B.; Fagin, J.A.; Solit, D.B.; Paty, P.B.; Rohle, D.; Vivanco, I.; Chmielecki, J.; et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc. Natl. Acad. Sci. USA 2009, 106, 9435–9440. [Google Scholar] [CrossRef] [Green Version]
- Krebs, D.L.; Hilton, D.J. SOCS Proteins: Negative Regulators of Cytokine Signaling. Stem Cells 2001, 19, 378–387. [Google Scholar] [CrossRef]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific inhibition of Stat3 signal transduction by PIAS3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K. Modulation of STAT signaling by STAT-interacting proteins. Oncogene 2000, 19, 2638–2644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, S.; Li, J.; Yu, L.; Sun, S. Protein tyrosine phosphatase 1B expression contributes to the development of breast cancer. J. Zhejiang Univ. B 2017, 18, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Banerjee, S. Differential PIAS3 expression in human malignancy. Oncol. Rep. 2004, 11, 1319–1324. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Oskooei, V.K.; Azari, I.; Taheri, M. Suppressor of cytokine signaling (SOCS) genes are downregulated in breast cancer. World J. Surg. Oncol. 2018, 16, 226. [Google Scholar] [CrossRef]
- Avalle, L.; Poli, V. Nucleus, Mitochondrion, or Reticulum? STAT3 à La Carte. Int. J. Mol. Sci. 2018, 19, 2820. [Google Scholar] [CrossRef] [Green Version]
- Gough, D.J.; Koetz, L.; Levy, D.E. The MEK-ERK pathway is necessary for serine phosphorylation of mitochondrial STAT3 and ras-mediated transformation. PLoS ONE 2013, 8, e83395. [Google Scholar] [CrossRef]
- Meier, J.A.; Hyun, M.; Cantwell, M.; Raza, A.; Mertens, C.; Raje, V.; Sisler, J.; Tracy, E.; Torres-Odio, S.; Gispert, S.; et al. Stress-induced dynamic regulation of mitochondrial STAT3 and its association with cyclophilin D reduce mitochondrial ROS production. Sci. Signal. 2017, 10, eaag2588. [Google Scholar] [CrossRef] [Green Version]
- Kurdi, M.; Booz, G.W. Evidence that IL-6-type cytokine signaling in cardiomyocytes is inhibited by oxidative stress: Parthenolide targets JAK1 activation by generating ROS. J. Cell. Physiol. 2007, 212, 424–431. [Google Scholar] [CrossRef]
- Xie, Y.; Kole, S.; Precht, P.; Pazin, M.J.; Bernier, M. S -Glutathionylation Impairs Signal Transducer and Activator of Transcription 3 Activation and Signaling. Endocrinology 2009, 150, 1122–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.S.; Liang, J.J.; Wang, Y.; Zhao, X.J.; Xu, L.; Xu, Y.; Zou, Q.C.; Zhang, J.M.; Tu, C.; Cui, Y.; et al. STAT3 Undergoes Acetylation-dependent Mitochondrial Translocation to Regulate Pyruvate Metabolism. Sci. Rep. 2016, 6, 39517. [Google Scholar] [CrossRef]
- Boengler, K.; Hilfiker-Kleiner, D.; Heusch, G.; Schulz, R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res. Cardiol. 2010, 105, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Ray, S.; Boldogh, I.; Brasier, A.R. STAT3 NH2-Terminal Acetylation Is Activated by the Hepatic Acute-Phase Response and Required for IL-6 Induction of Angiotensinogen. Gastroenterology 2005, 129, 1616–1632. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zhang, P.; Herrmann, A.; Yang, C.; Xin, H.; Wang, Z.; Hoon, D.S.B.; Forman, S.J.; Jove, R.; Riggs, A.D.; et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. USA 2012, 109, 7765–7769. [Google Scholar] [CrossRef] [Green Version]
- Tammineni, P.; Anugula, C.; Mohammed, F.; Anjaneyulu, M.; Larner, A.C.; Sepuri, N.B.V.; Babu, N.; Sepuri, V. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J. Biol. Chem. 2013, 288, 4723–4732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rincon, M.; Pereira, F.V. A new perspective: Mitochondrial stat3 as a regulator for lymphocyte function. Int. J. Mol. Sci. 2018, 19, 1656. [Google Scholar] [CrossRef] [Green Version]
- Shulga, N.; Pastorino, J.G. GRIM-19-mediated translocation of STAT3 to mitochondria is necessary for TNF-induced necroptosis. J. Cell Sci. 2012, 125, 2995–3003. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Rincon, M. Mitochondrial Stat3, the Need for Design Thinking. Int. J. Biol. Sci. 2016, 12, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 Supports Ras-Dependent Oncogenic Transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Sheng, M.; Wang, J.; Teng, T.; Sun, Y.; Yang, Q.; Xu, Z. Zinc improves mitochondrial respiratory function and prevents mitochondrial ROS generation at reperfusion by phosphorylating STAT3 at Ser727. J. Mol. Cell. Cardiol. 2018, 118, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Poli, V.; Camporeale, A. STAT3-Mediated Metabolic Reprograming in Cellular Transformation and Implications for Drug Resistance. Front. Oncol. 2015, 5, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Raje, V.; Yakovlev, V.A.; Yacoub, A.; Szczepanek, K.; Meier, J.; Derecka, M.; Chen, Q.; Hu, Y.; Sisler, J.; et al. Mitochondrial Localized Stat3 Promotes Breast Cancer Growth via Phosphorylation of Serine 727. J. Biol. Chem. 2013, 288, 31280–31288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Too, H.-P. Mitochondrial Localized STAT3 Is Involved in NGF Induced Neurite Outgrowth. PLoS ONE 2011, 6, e21680. [Google Scholar] [CrossRef]
- Meier, J.A.; Larner, A.C. Toward a new STATe: The role of STATs in mitochondrial function. Semin. Immunol. 2014, 26, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Sarafian, T.A.; Montes, C.; Imura, T.; Qi, J.; Coppola, G.; Geschwind, D.H.; Sofroniew, M.V. Disruption of Astrocyte STAT3 Signaling Decreases Mitochondrial Function and Increases Oxidative Stress In Vitro. PLoS ONE 2010, 5, e9532. [Google Scholar] [CrossRef] [Green Version]
- Kramer, A.H.; Edkins, A.L.; Hoppe, H.C.; Prinsloo, E. Dynamic mitochondrial localisation of STAT3 in the cellular adipogenesis model 3T3-L1. J. Cell. Biochem. 2015, 116, 1232–1240. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Cai, T.; Kuang, Y.; Zhang, C.; Zhang, Z.; Chen, L.; Li, B.; Li, Y.; Wang, Y.; Yang, H.; Han, Q.; et al. Glucose-6-phosphate dehydrogenase and NADPH oxidase 4 control STAT3 activity in melanoma cells through a pathway involving reactive oxygen species, c-SRC and SHP2. Am. J. Cancer Res. 2015, 5, 1610–1620. [Google Scholar]
- Mauer, J.; Denson, J.L.; Brüning, J.C. Versatile functions for IL-6 in metabolism and cancer. Trends Immunol. 2015, 36, 92–101. [Google Scholar] [CrossRef]
- Doerstling, S.S.; O’Flanagan, C.H.; Hursting, S.D. Obesity and Cancer Metabolism: A Perspective on Interacting Tumor–Intrinsic and Extrinsic Factors. Front. Oncol. 2017, 7, 216. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Aiderus, A.; Black, M.A.; Dunbier, A.K. Fatty acid oxidation is associated with proliferation and prognosis in breast and other cancers. BMC Cancer 2018, 18, 805. [Google Scholar] [CrossRef]
- Luo, X.; Zhao, X.; Cheng, C.; Li, N.; Liu, Y.; Cao, Y. The implications of signaling lipids in cancer metastasis. Exp. Mol. Med. 2018, 50, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louie, S.M.; Roberts, L.S.; Mulvihill, M.M.; Luo, K.; Nomura, D.K. Cancer cells incorporate and remodel exogenous palmitate into structural and oncogenic signaling lipids. Biochim. Biophys. Acta 2013, 1831, 1566–1572. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.J.; Tripathi, S.C.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y.; et al. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 2018, 27, 136.e5–150.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Yue, C.; Herrmann, A.; Song, J.; Egelston, C.; Wang, T.; Zhang, Z.; Li, W.; Lee, H.; Aftabizadeh, M.; et al. STAT3 Activation-Induced Fatty Acid Oxidation in CD8+ T Effector Cells Is Critical for Obesity-Promoted Breast Tumor Growth. Cell Metab. 2020, 31, 148.e5–161.e5. [Google Scholar] [CrossRef] [PubMed]
- Fiorenza, L.; Yong, R.; Ranjitkar, S.; Hughes, T.; Quayle, M.; McMenamin, P.G.; Kaidonis, J.; Townsend, G.C.; Adams, J.W. Technical note: The use of 3D printing in dental anthropology collections. Am. J. Phys. Anthropol. 2018, 167, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Staniszewska, A.D.; Pensa, S.; Caffarel, M.M.; Anderson, L.H.; Poli, V.; Watson, C.J. Stat3 Is Required to Maintain the Full Differentiation Potential of Mammary Stem Cells and the Proliferative Potential of Mammary Luminal Progenitors. PLoS ONE 2012, 7, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Keymeulen, A.; Rocha, A.S.; Ousset, M.; Beck, B.; Bouvencourt, G.; Rock, J.; Sharma, N.; Dekoninck, S.; Blanpain, C. Distinct stem cells contribute to mammary gland development and maintenance. Nature 2011, 479, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Rios, A.C.; Fu, N.Y.; Lindeman, G.J.; Visvader, J.E. In situ identification of bipotent stem cells in the mammary gland. Nature 2014, 506, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Brenot, A.; Lawson, D.A.; Linnemann, J.R.; Van Kappel, E.C.; Wong, K.C.; de Sauvage, F.; Klein, O.D.; Werb, Z. Lgr5-Expressing Cells Are Sufficient And Necessary for Postnatal Mammary Gland Organogenesis. Cell Rep. 2013, 3, 70–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Dontu, G.; Wicha, M.S. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005, 7, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Tang, H.; Kong, Y.; Xie, X.; Chen, J.; Song, C.; Liu, X.; Ye, F.; Li, N.; Wang, N.; et al. LGR5 promotes breast cancer progression and maintains stem-like cells through activation of wnt/β-catenin signaling. Stem Cells 2015, 33, 2913–2924. [Google Scholar] [CrossRef]
- Yang, F.; Xu, J.; Tang, L.; Guan, X. Breast cancer stem cell: The roles and therapeutic implications. Cell. Mol. Life Sci. 2017, 74, 951–966. [Google Scholar] [CrossRef]
- Jia, J.; Shi, Y.; Yan, B.; Xiao, D.; Lai, W.; Pan, Y.; Jiang, Y.; Chen, L.; Mao, C.; Zhou, J.; et al. LGR5 expression is controled by IKKα in basal cell carcinoma through activating STAT3 signaling pathway. Oncotarget 2016, 7, 27280–27294. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Garama, D.J.; White, C.L.; Balic, J.J.; Gough, D.J. Mitochondrial STAT3: Powering up a potent factor. Cytokine 2016, 87, 20–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, E.G.; Cotter, T.G. New Insight into the Role of Reactive Oxygen Species (ROS) in Cellular Signal-Transduction Processes. Int. Rev. Cell Mol. Biol. 2015, 319, 221–254. [Google Scholar] [PubMed]
- Kuang, X.; Xiong, J.; Wang, W.; Li, X.; Lu, T.; Fang, Q.; Wang, J. PIM inhibitor SMI-4a induces cell apoptosis in B-cell acute lymphocytic leukemia cells via the HO-1-mediated JAK2/STAT3 pathway. Life Sci. 2019, 219, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Avagliano, A.; Ruocco, M.R.; Aliotta, F.; Belviso, I.; Accurso, A.; Masone, S.; Montagnani, S.; Arcucci, A. Mitochondrial Flexibility of Breast Cancers: A Growth Advantage and a Therapeutic Opportunity. Cells 2019, 8, 401. [Google Scholar] [CrossRef] [Green Version]
- Cozzo, A.J.; Fuller, A.M.; Makowski, L. Contribution of Adipose Tissue to Development of Cancer. Compr. Physiol. 2017, 8, 237–282. [Google Scholar] [PubMed]
- Catz, S.D.; Johnson, J.L. BCL-2 in prostate cancer: A minireview. Apoptosis 2003, 8, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Findley, H.W.; Gu, L.; Yeager, A.M.; Zhou, M. Expression and regulation of Bcl-2, Bcl-xl, and Bax correlate with p53 status and sensitivity to apoptosis in childhood acute lymphoblastic leukemia. Blood 1997, 89, 2986–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curry, M.C.; Luk, N.A.; Kenny, P.A.; Roberts-Thomson, S.J.; Monteith, G.R. Distinct Regulation of Cytoplasmic Calcium Signals and Cell Death Pathways by Different Plasma Membrane Calcium ATPase Isoforms in MDA-MB-231 Breast Cancer Cells. J. Biol. Chem. 2012, 287, 28598–28608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kania, E.; Pająk, B.; Orzechowski, A. Calcium Homeostasis and ER Stress in Control of Autophagy in Cancer Cells. Biomed. Res. Int. 2015, 2015, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Liu, Y.; Song, F.; Zheng, H.; Hu, L.; Lu, H.; Liu, P.; Hao, X.; Zhang, W.; Chen, K. Functional SNP in the microRNA-367 binding site in the 3’UTR of the calcium channel ryanodine receptor gene 3 (RYR3) affects breast cancer risk and calcification. Proc. Natl. Acad. Sci. USA 2011, 108, 13653–13658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Shen, Q.; Claret, F.X. Novel roles of reactive oxygen species in the pathogenesis of acute myeloid leukemia. J. Leukoc. Biol. 2013, 94, 423–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissman, H.M.; Hayek, B.R.; Grossniklaus, H.E. Reticulohistiocytoma of the Orbit. Ophthalmic Plast. Reconstr. Surg. 2015, 31, e13–e16. [Google Scholar] [CrossRef] [PubMed]
- Matsui, A.; Ikeda, T.; Enomoto, K.; Hosoda, K.; Nakashima, H.; Omae, K.; Watanabe, M.; Hibi, T.; Kitajima, M. Increased formation of oxidative DNA damage, 8-hydroxy-2’- deoxyguanosine, in human breast cancer tissue and its relationship to GSTP1 and COMT genotypes. Cancer Lett. 2000, 151, 87–95. [Google Scholar] [CrossRef]
- Okoh, V.; Deoraj, A.; Roy, D. Estrogen-induced reactive oxygen species-mediated signalings contribute to breast cancer. Biochim. Biophys. Acta 2011, 1815, 115–133. [Google Scholar] [CrossRef]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Vitale, I.; Rigoni, A.; Vacchelli, E.; Michaud, M.; Zischka, H.; Castedo, M.; Kroemer, G. Mitochondrial gateways to cancer. Mol. Aspects Med. 2010, 31, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial Membrane Permeabilization in Cell Death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Wu, Q.; Ni, X. ROS-Mediated DNA Methylation Pattern Alterations in Carcinogenesis. Curr. Drug Targets 2015, 16, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Agirre, X.; Vizmanos, J.L.; Calasanz, M.J.; García-Delgado, M.; Larráyoz, M.J.; Novo, F.J. Methylation of CpG dinucleotides and/or CCWGG motifs at the promoter of TP53 correlates with decreased gene expression in a subset of acute lymphoblastic leukemia patients. Oncogene 2003, 22, 1070–1072. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.T.; Su, J.C.; Liu, C.Y.; Shiau, C.W.; Chen, K.F. Alteration of SHP-1/p-STAT3 signaling: A potential target for anticancer therapy. Int. J. Mol. Sci. 2017, 18, 1234. [Google Scholar] [CrossRef] [Green Version]
- Salganik, R.I. The Benefits and Hazards of Antioxidants: Controlling Apoptosis and Other Protective Mechanisms in Cancer Patients and the Human Population. J. Am. Coll. Nutr. 2001, 20, 464S–472S. [Google Scholar] [CrossRef] [PubMed]
- Kamata, H.; Hirata, H. Redox regulation of cellular signalling. Cell Signal. 1999, 11, 1–14. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Ng, I.H.W.; Yeap, Y.Y.C.; Ong, L.S.R.; Jans, D.A.; Bogoyevitch, M.A. Oxidative stress impairs multiple regulatory events to drive persistent cytokine-stimulated STAT3 phosphorylation. Biochim. Biophys. Acta 2014, 1843, 483–494. [Google Scholar] [CrossRef] [Green Version]
- Tormos, K.V.V.T.; Anso, E.; Hanamaka, R.H.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.; Chandel, N.S.S. Mitochondrial Complex III ROS Regulate Adipocyte Differentiation. Cell Metab. 2011, 14, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Jankovic, A.; Korac, A.; Srdic-Galic, B.; Buzadzic, B.; Otasevic, V.; Stancic, A.; Vucetic, M.; Markelic, M.; Velickovic, K.; Golic, I.; et al. Differences in the redox status of human visceral and subcutaneous adipose tissues – relationships to obesity and metabolic risk. Metabolism 2014, 63, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Wang, Z.; Li, H.; Shou, J.; Jing, Z.; Xie, J.; Sui, X.; Pan, H.; Han, W. The role of STAT3 in autophagy. Autophagy 2015, 11, 729–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyamfi, J.; Eom, M.; Koo, J.S.; Choi, J. Multifaceted Roles of Interleukin-6 in Adipocyte–Breast Cancer Cell Interaction. Transl. Oncol. 2018, 11, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Coqueret, O.; Bélanger, A.; Barré, B.; Guette, C.; Jonchère, B. STAT3 as a new autophagy regulator. JAKSTAT 2013, 2, e24353. [Google Scholar]
- Gyamfi, J.; Lee, Y.H.; Eom, M.; Choi, J. Interleukin-6/STAT3 signalling regulates adipocyte induced epithelial-mesenchymal transition in breast cancer cells. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Maycotte, P.; Gearheart, C.M.; Barnard, R.; Aryal, S.; Mulcahy Levy, J.M.; Fosmire, S.P.; Hansen, R.J.; Morgan, M.J.; Porter, C.C.; Gustafson, D.L.; et al. STAT3-Mediated Autophagy Dependence Identifies Subtypes of Breast Cancer Where Autophagy Inhibition Can Be Efficacious. Cancer Res. 2014, 74, 2579–2590. [Google Scholar] [CrossRef] [Green Version]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef]
- Akhtar, F.; Bokhari, S.R.A. Apoptosis; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Seo, H.-S.; Jo, J.K.; Ku, J.M.; Choi, H.-S.; Choi, Y.K.; Woo, J.-K.; Kim, H.I.; Kang, S.Y.; Lee, K.M.; Nam, K.W.; et al. Induction of caspase-dependent extrinsic apoptosis by apigenin through inhibition of signal transducer and activator of transcription 3 (STAT3) signalling in HER2-overexpressing BT-474 breast cancer cells. Biosci. Rep. 2015, 35, e00276. [Google Scholar] [CrossRef] [Green Version]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Shimizu, T.; Numata, T.; Okada, Y. A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl(-) channel. Proc. Natl. Acad. Sci. USA 2004, 101, 6770–6773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.G.; Lai, P.B.S. Apoptosis in Carcinogenesis and Chemotherapy; Springer: Dordrecht, The Netherlands, 2009; pp. 1–384. [Google Scholar]
- Punnoose, E.A.; Leverson, J.D.; Peale, F.; Boghaert, E.R.; Belmont, L.D.; Tan, N.; Young, A.; Mitten, M.; Ingalla, E.; Darbonne, W.C.; et al. Expression Profile of BCL-2, BCL-XL, and MCL-1 Predicts Pharmacological Response to the BCL-2 Selective Antagonist Venetoclax in Multiple Myeloma Models. Mol. Cancer Ther. 2016, 15, 1132–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodapasand, E.; Jafarzadeh, N.; Farrokhi, F.; Kamalidehghan, B.; Houshmand, M. Is Bax/Bcl-2 ratio considered as a prognostic marker with age and tumor location in colorectal cancer? Iran. Biomed. J. 2015, 19, 69–75. [Google Scholar] [PubMed]
- Turkson, J.; Jove, R. STAT proteins: Novel molecular targets for cancer drug discovery. Oncogene 2000, 19, 6613–6626. [Google Scholar] [CrossRef] [Green Version]
- Rozovski, U.; Harris, D.M.; Li, P.; Liu, Z.; Wu, J.Y.; Grgurevic, S.; Faderl, S.; Ferrajoli, A.; Wierda, W.G.; Martinez, M.; et al. At High Levels, Constitutively Activated STAT3 Induces Apoptosis of Chronic Lymphocytic Leukemia Cells. J. Immunol. 2016, 196, 4400–4409. [Google Scholar] [CrossRef]
- Jackson, N.M.; Ceresa, B.P. EGFR-mediated apoptosis via STAT3. Exp. Cell Res. 2017, 356, 93–103. [Google Scholar] [CrossRef]
- Yang, R.; Lirussi, D.; Thornton, T.M.; Jelley-Gibbs, D.M.; Diehl, S.A.; Case, L.K.; Madesh, M.; Taatjes, D.J.; Teuscher, C.; Haynes, L.; et al. Mitochondrial Ca2+ and membrane potential, an alternative pathway for Interleukin 6 to regulate CD4 cell effector function. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Hui, K.; Hu, C.; Wen, Y.; Yang, S.; Zhu, P.; Wang, L.; Xia, Y. Autophagy inhibition potentiates the anti- angiogenic property of multikinase inhibitor anlotinib through JAK2/STAT3/VEGFA signaling in non-small cell lung cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 71. [Google Scholar] [CrossRef]
- Held, N.M.; Houtkooper, R.H. Mitochondrial quality control pathways as determinants of metabolic health. BioEssays 2015, 37, 867–876. [Google Scholar] [CrossRef]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer 2015, 14, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boya, P.; Codogno, P.; Rodriguez-Muela, N. Autophagy in stem cells: Repair, remodelling and metabolic reprogramming. Development 2018, 145, dev146506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Lluch, G. Mitochondrial activity and dynamics changes regarding metabolism in ageing and obesity. Mech. Ageing Dev. 2017, 162, 108–121. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Yang, Y.; Ming, M.; Liu, B. Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochem. Biophys. Res. Commun. 2011, 414, 5–8. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics—Fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344. [Google Scholar] [CrossRef]
- Niso-Santano, M.; Shen, S.; Adjemian, S.; Malik, S.A.; Mariño, G.; Lachkar, S.; Senovilla, L.; Kepp, O.; Galluzzi, L.; Maiuri, M.C.; et al. Direct interaction between STAT3 and EIF2AK2 controls fatty acid-induced autophagy. Autophagy 2013, 9, 415–417. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Niso-Santano, M.; Adjemian, S.; Takehara, T.; Malik, S.A.; Minoux, H.; Souquere, S.; Mariño, G.; Lachkar, S.; Senovilla, L.; et al. Cytoplasmic STAT3 Represses Autophagy by Inhibiting PKR Activity. Mol. Cell 2012, 48, 667–680. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Loux, T.; Tang, D.; Schapiro, N.E.; Vernon, P.; Livesey, K.M.; Krasinskas, A.; Lotze, M.T.; Zeh, H.J. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc. Natl. Acad. Sci. USA 2012, 109, 7031–7036. [Google Scholar] [CrossRef] [Green Version]
- Van Kruijsdijk, R.C.M.; van der Wall, E.; Visseren, F.L.J. Obesity and Cancer: The Role of Dysfunctional Adipose Tissue. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2569–2578. [Google Scholar] [CrossRef] [Green Version]
- Divella, R.; De Luca, R.; Abbate, I.; Naglieri, E.; Daniele, A. Obesity and cancer: The role of adipose tissue and adipo-cytokines-induced chronic inflammation. J. Cancer 2016, 7, 2346–2359. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [Green Version]
- Lauby-Secretan, B.; Straif, K.; Scoccianti, C.; Grosse, Y.; Bianchini, F.; Loomis, D. Body Fatness and Cancer —Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.; Wang, Y.; Peng, W.; Xu, L.; Liu, M.; Li, J.; Hu, X.; Li, Y.; Zuo, J.; Ye, Y. STAT3 activation by IL-6 from adipose-derived stem cells promotes endometrial carcinoma proliferation and metastasis. Biochem. Biophys. Res. Commun. 2018, 500, 626–631. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Appari, M.; Channon, K.M.; McNeill, E. Metabolic Regulation of Adipose Tissue Macrophage Function in Obesity and Diabetes. Antioxid. Redox Signal. 2017, 29, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Kratz, M.; Coats, B.R.; Hisert, K.B.; Hagman, D.; Mutskov, V.; Peris, E.; Schoenfelt, K.Q.; Kuzma, J.N.; Larson, I.; Billing, P.S.; et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014, 20, 614–625. [Google Scholar] [CrossRef] [Green Version]
- Takagi, M.; Uno, H.; Nishi, R.; Sugimoto, M.; Hasegawa, S.; Piao, J.; Ihara, N.; Kanai, S.; Kakei, S.; Tamura, Y.; et al. ATM Regulates Adipocyte Differentiation and Contributes to Glucose Homeostasis. Cell Rep. 2015, 10, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Corrêa, L.H.; Corrêa, R.; Farinasso, C.M.; de Sant’Ana Dourado, L.P.; Magalhães, K.G. Adipocytes and macrophages interplay in the orchestration of tumor microenvironment: New implications in cancer progression. Front. Immunol. 2017, 8, 1129. [Google Scholar] [CrossRef]
- Kuroda, M.; Sakaue, H. Adipocyte Death and Chronic Inflammation in Obesity. J. Med. Investig. 2017, 64, 193–196. [Google Scholar] [CrossRef] [Green Version]
- Fischer-Posovszky, P.; Wang, Q.A.; Asterholm, I.W.; Rutkowski, J.M.; Scherer, P.E. Targeted Deletion of Adipocytes by Apoptosis Leads to Adipose Tissue Recruitment of Alternatively Activated M2 Macrophages. Endocrinology 2011, 152, 3074–3081. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.A.; Parker, E.; Blair, C.K.; Cerhan, J.R.; Folsom, A.R. Body mass index and risk of leukemia in older women. Cancer Epidemiol. Biomark. Prev. 2004, 13, 1810–1813. [Google Scholar] [CrossRef] [PubMed]
- Brandon, E.L.; Gu, J.W.; Cantwell, L.; He, Z.; Wallace, G.; Hall, J.E. Obesity promotes melanoma tumor growth: Role of leptin. Cancer Biol. Ther. 2009, 8, 1871–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapp, K.; Schroeder, J.; Klenk, J.; Stoehr, S.; Ulmer, H.; Concin, H.; Diem, G.; Oberaigner, W.; Weiland, S.K. Obesity and incidence of cancer: A large cohort study of over 145,000 adults in Austria. Br. J. Cancer 2005, 93, 1062–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Demaria, M.; Camporeale, A.; Poli, V. STAT3 and metabolism: How many ways to use a single molecule? Int. J. Cancer 2014, 135, 1997–2003. [Google Scholar] [CrossRef]
- Binai, N.A.; Damert, A.; Carra, G.; Steckelbroeck, S.; Löwer, J.; Löwer, R.; Wessler, S. Expression of estrogen receptor alpha increases leptin-induced STAT3 activity in breast cancer cells. Int. J. Cancer 2010, 127, 55–66. [Google Scholar] [CrossRef]
- Bar-Natan, M.; Nelson, E.A.; Xiang, M.; Frank, D.A. STAT signaling in the pathogenesis and treatment of myeloid malignancies. JAKSTAT 2012, 1, 55–64. [Google Scholar] [CrossRef]
- Bougaret, L.; Delort, L.; Billard, H.; Le Huede, C.; Boby, C.; De la Foye, A.; Rossary, A.; Mojallal, A.; Damour, O.; Auxenfans, C.; et al. Adipocyte/breast cancer cell crosstalk in obesity interferes with the anti-proliferative efficacy of tamoxifen. PLoS ONE 2018, 13, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.H.; Wang, P.J.; Hsieh, Y.C.; Lo, S.; Lee, Y.C.; Chen, Y.C.; Tsai, C.H.; Chiu, W.C.; Hu, S.C.S.; Lu, C.W.; et al. Resistin facilitates breast cancer progression via TLR4- mediated induction of mesenchymal phenotypes and stemness properties. Oncogene 2018, 37, 589–600. [Google Scholar] [CrossRef]
- Chen, S.H.; Murphy, D.A.; Lassoued, W.; Thurston, G.; Feldman, M.D.; Lee, W.M.F. Activated STAT3 is a mediator and biomarker of VEGF endothelial activation. Cancer Biol. Ther. 2008, 7, 1994–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, A.; Ramalho-Santos, J.; St John, J.C. The expression of polymerase gamma and mitochondrial transcription factor A and the regulation of mitochondrial DNA content in mature human sperm. Hum. Reprod. 2007, 22, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Schäfer, H. Role of the immediate early response 3 (IER3) gene in cellular stress response, inflammation and tumorigenesis. Eur. J. Cell Biol. 2011, 90, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Shen, Y.; Yang, J.; Li, S.; Wang, B.; Chen, Z.; Li, P.; Liu, P.; Yang, J. Angiomotin Family Members: Oncogenes or Tumor Suppressors? Int. J. Biol. Sci. 2017, 13, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Cairns, J.; Fridley, B.L.; Jenkins, G.D.; Zhuang, Y.; Yu, J.; Wang, L. Differential roles of ERRFI1 in EGFR and AKT pathway regulation affect cancer proliferation. EMBO Rep. 2018, 19, e44767. [Google Scholar] [CrossRef]
- Fox, S.B.; Bragança, J.; Turley, H.; Campo, L.; Han, C.; Gatter, K.C.; Bhattacharya, S.; Harris, A.L. CITED4 inhibits hypoxia-activated transcription in cancer cells, and its cytoplasmic location in breast cancer is associated with elevated expression of tumor cell hypoxia-inducible factor 1α. Cancer Res. 2004, 64, 6075–6081. [Google Scholar] [CrossRef] [Green Version]
- Begicevic, R.R.; Falasca, M. ABC transporters in cancer stem cells: Beyond chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [Green Version]
- Kristiansen, M.; Langer, A.; Knudsen, G.; Weber, B.; Børresen-Dale, A.-L.; Ørstavik, K. X chromosome inactivation pattern in female patients with breast cancer. Breast Cancer Res. 2000, 2, P1–P10. [Google Scholar] [CrossRef]
- Pau, C.T.; Mosbruger, T.; Saxena, R.; Welt, C.K. Phenotype and tissue expression as a function of genetic risk in polycystic ovary syndrome. PLoS ONE 2017, 12, 168870. [Google Scholar] [CrossRef]
- Grimshaw, M.J.; Hagemann, T.; Ayhan, A.; Gillett, C.E.; Binder, C.; Balkwill, F.R. A Role for Endothelin-2 and Its Receptors in Breast Tumor Cell Invasion. Cancer Res. 2004, 64, 2461–2468. [Google Scholar] [CrossRef] [Green Version]
- Kumarswamy, R.; Volkmann, I.; Thum, T. Regulation and function of miRNA-21 in health and disease. RNA Biol. 2011, 8, 706–713. [Google Scholar] [CrossRef] [Green Version]
- Peralta-Zaragoza, O.; Deas, J.; Meneses-Acosta, A.; De la O-Gómez, F.; Fernández-Tilapa, G.; Gómez-Cerón, C.; Benítez-Boijseauneau, O.; Burguete-García, A.; Torres-Poveda, K.; Bermúdez-Morales, V.H.; et al. Relevance of miR-21 in regulation of tumor suppressor gene PTEN in human cervical cancer cells. BMC Cancer 2016, 16, 215. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhang, B.; Bao, L.; Jin, L.; Yang, M.; Peng, Y.; Kumar, A.; Wang, J.E.; Wang, C.; Zou, X.; et al. ZMYND8 acetylation mediates HIF-dependent breast cancer progression and metastasis. J. Clin. Investig. 2018, 128, 1937–1955. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Miller, K.M. Double duty: ZMYND8 in the DNA damage response and cancer. Cell Cycle 2018, 17, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Frey, S.; Riethdorf, S.; Schulze, C.; Alawi, M.; Kling, L.; Vafaizadeh, V.; Sauter, G.; Terracciano, L.; Schumacher, U.; et al. Dual roles of the transcription factor grainyhead-like 2 (GRHL2) in breast cancer. J. Biol. Chem. 2013, 288, 22993–23008. [Google Scholar] [CrossRef] [Green Version]
- Cunliffe, H.E.; Jiang, Y.; Fornace, K.M.; Yang, F.; Meltzer, P.S. PAR6B is required for tight junction formation and activated PKCζ localization in breast cancer. Am. J. Cancer Res. 2012, 2, 478–491. [Google Scholar]
- Liu, X.; Bi, L.; Wang, Q.; Wen, M.; Li, C.; Ren, Y.; Jiao, Q.; Mao, J.H.; Wang, C.; Wei, G.; et al. MiR-1204 targets VDR to promotes epithelial-mesenchymal transition and metastasis in breast cancer. Oncogene 2018, 37, 3426–3439. [Google Scholar] [CrossRef] [Green Version]
- Hamada, T.; Souda, M.; Yoshimura, T.; Sasaguri, S.; Hatanaka, K.; Tasaki, T.; Yoshioka, T.; Ohi, Y.; Yamada, S.; Tsutsui, M.; et al. Anti-apoptotic Effects of PCP4/PEP19 in Human Breast Cancer Cell Lines: A Novel Oncotarget. Oncotarget 2014, 5, 6076–6086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, T.; Hamada, T.; Hijioka, H.; Souda, M.; Hatanaka, K.; Yoshioka, T.; Yamada, S.; Tsutsui, M.; Umekita, Y.; Nakamura, N.; et al. PCP4/PEP19 promotes migration, invasion and adhesion in human breast cancer MCF-7 and T47D cells. Oncotarget 2016, 7, 49065–49074. [Google Scholar] [CrossRef] [Green Version]
- Thakkar, A.; Raj, H.; Ravishankar; Muthuvelan, B.; Balakrishnan, A.; Padigaru, M. High expression of three-gene signature improves prediction of relapse-free survival in estrogen receptor-positive and node-positive breast tumors. Biomark. Insights 2015, 10, 103–112. [Google Scholar] [CrossRef]
- Jiang, Y.; Qian, F.; Bai, X.; Liu, Y.; Wang, Q.; Ai, B.; Han, X.; Shi, S.; Zhang, J.; Li, X.; et al. SEdb: A comprehensive human super-enhancer database. Nucleic Acids Res. 2019, 47, D235–D243. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.C.; Li, X.C.; Guo, J.C.; Zhao, J.M.; Li, Y.Y.; Tang, Z.D.; Zhou, L.W.; Zhang, J.; Bai, X.F.; Jiang, Y.; et al. SEanalysis: A web tool for super-enhancer associated regulatory analysis. Nucleic Acids Res. 2019, 47, W248–W255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esparza, E.M.; Arch, R.H. TRAF4 functions as an intermediate of GITR-induced NF-κB activation. Cell. Mol. Life Sci. 2004, 61, 3087–3092. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Mao, R.; Yang, J. NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandard, S.; Müller, M.; Kersten, S. Peroxisome Proliferator-Activated Receptor Alpha Target Genes. Cell. Mol. Life Sci. 2004, 61, 393–416. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Liu, Y.; Wang, J.; Yuan, X.; Jin, H.W.; Zhang, L.R.; Zhang, J.T.; Liu, Z.M.; Cui, J.R. Small-molecule compounds targeting the STAT3 DNA-binding domain suppress survival of cisplatin-resistant human ovarian cancer cells by inducing apoptosis. Eur. J. Med. Chem. 2018, 157, 887–897. [Google Scholar] [CrossRef] [Green Version]
- Mociño-Rodríguez, M.D.; Santillán-Benítez, J.G.; Dozal-Domínguez, D.S.; Hernández-Navarro, M.D.; Flores-Merino, M.V.; Sandoval-Cabrera, A.; García Vázquez, F.J. Expression of AdipoR1 and AdipoR2 Receptors as Leptin-Breast Cancer Regulation Mechanisms. Dis. Markers 2017, 2017, 1–11. [Google Scholar] [CrossRef]
- Dalamaga, M.; Diakopoulos, K.N.; Mantzoros, C.S. The Role of Adiponectin in Cancer: A Review of Current Evidence. Endocr. Rev. 2012, 33, 547–594. [Google Scholar] [CrossRef] [Green Version]
- Katira, A.; Tan, P.H. Evolving role of adiponectin in cancer-controversies and update. Cancer Biol. Med. 2016, 13, 101–119. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| STAT3 Activators | Cell Lines | |
|---|---|---|
| Cytokines | IL-6 | Prostate cancer, pancreatic cancer, macrophages [56,57,58] |
| IL-10 | Chronic lymphocytic leukemia, macrophages [59] | |
| IL-17 | Hepatocellular carcinoma, stromal cells [60] | |
| Interferons | Lung fibrosarcoma [61] | |
| Hormones | Leptin | Hipothalamus [54] |
| Growth factors | Granulocyte colony-stimulating factor | Bone marrow neutrophils, monocytes [62,63] |
| Epidermal growth factor | In vitro [64,65,66] | |
| Platelet-derived growth factor | 3T3 cells and fibroblasts [65,67] | |
| Oncogenes | Src | Fibroblasts, glioblastoma [68,69] |
| Rac1 | COS-1 fibroblasts [65] | |
| Bone marrow X-linked nonreceptor tyrosine kinase | Glioblastoma [68] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadye, R.; Stoffels, M.; Fanucci, S.; Mbanxa, S.; Prinsloo, E. A STAT3 of Addiction: Adipose Tissue, Adipocytokine Signalling and STAT3 as Mediators of Metabolic Remodelling in the Tumour Microenvironment. Cells 2020, 9, 1043. https://doi.org/10.3390/cells9041043
Kadye R, Stoffels M, Fanucci S, Mbanxa S, Prinsloo E. A STAT3 of Addiction: Adipose Tissue, Adipocytokine Signalling and STAT3 as Mediators of Metabolic Remodelling in the Tumour Microenvironment. Cells. 2020; 9(4):1043. https://doi.org/10.3390/cells9041043
Chicago/Turabian StyleKadye, Rose, Mihlali Stoffels, Sidne Fanucci, Siso Mbanxa, and Earl Prinsloo. 2020. "A STAT3 of Addiction: Adipose Tissue, Adipocytokine Signalling and STAT3 as Mediators of Metabolic Remodelling in the Tumour Microenvironment" Cells 9, no. 4: 1043. https://doi.org/10.3390/cells9041043