Exploring the Relevance of Senotherapeutics for the Current SARS-CoV-2 Emergency and Similar Future Global Health Threats

 , ,
, ,  , and
, and
Abstract
:1. Introduction
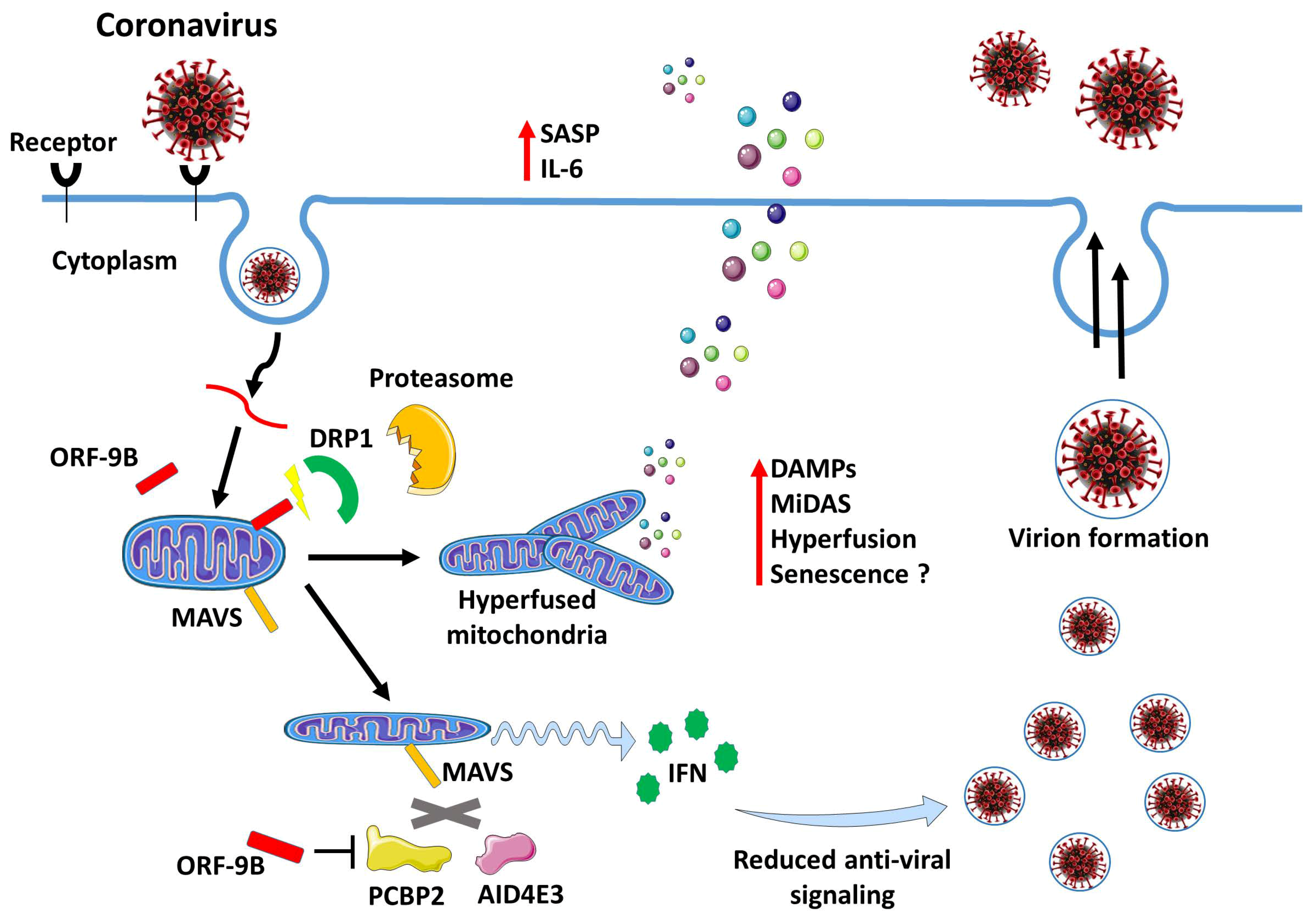
2. Mechanism Driving Inflammation during SARS-CoV-2 Infection
3. Cellular Senescence and Inflammatory Response
4. Cellular Senescence and Response to Viruses
5. Therapeutic Perspectives
Funding
Conflicts of Interest
References
- Chan-Yeung, M.; Xu, R.H. SARS: Epidemiology. Respirology 2003, 8, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Jung, S.; Linton, N.M.; Kinoshita, R.; Hayashi, K.; Miyama, T.; Anzai, A.; Yang, Y.; Yuan, B.; Akhmetzhanov, A.R.; et al. Communicating the Risk of Death from Novel Coronavirus Disease (COVID-19). J. Clin. Med. 2020, 9, 580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Chen, Y.; Lin, R.; Han, K. Clinical feature of COVID-19 in elderly patients: A comparison with young and middle-aged patients. J. Infect. 2020. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of the SARS-CoV-2 by full-length human ACE2. Science 2020. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.-R.; Cao, Q.-D.; Hong, Z.-S.; Tan, Y.-Y.; Chen, S.-D.; Jin, H.-J.; Tan, K.-S.; Wang, D.-Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak – an update on the status. Mil. Med. Res. 2020, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elshahat, M.E.; Elfiky, A.A. COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 2020. pii: S0163-4453(20)30107-9. [Google Scholar] [CrossRef]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 2020, 176. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Conti, P.; Ronconi, G.; Caraffa, A.; Gallenga, C.; Ross, R.; Frydas, I.; Kritas, S. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by COVID-19: Anti-inflammatory strategies. J. Biol. Regul. Homeost. Agents 2020, 34. [Google Scholar]
- Zampino, M.; Ferrucci, L.; Semba, R.D. Biomarkers in the path from cellular senescence to frailty. Exp. Gerontol. 2020, 129, 110750. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Yu, Z.; Sunchu, B.; Shoaf, J.; Dang, I.; Zhao, S.; Caples, K.; Bradley, L.; Beaver, L.M.; Ho, E.; et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell 2017, 16, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Wang, X.; Geng, P.; Tang, X.; Xiang, L.; Lu, X.; Li, J.; Ruan, Z.; Chen, J.; Xie, G.; et al. Melatonin regulates PARP1 to control the senescence-associated secretory phenotype (SASP) in human fetal lung fibroblast cells. J. Pineal Res. 2017, 63. [Google Scholar] [CrossRef]
- Stebbing, J.; Phelan, A.; Griffin, I.; Tucker, C.; Oechsle, O.; Smith, D.; Richardson, P. COVID-19: Combining antiviral and anti-inflammatory treatments. Lancet Infect. Dis. 2020, 20, 400–402. [Google Scholar] [CrossRef]
- Kim, E.C.; Kim, J.R. Senotherapeutics: Emerging strategy for healthy aging and age-related disease. BMB Rep. 2019, 52, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Palmer, A.K.; Ding, H.; Weivoda, M.M.; Pirtskhalava, T.; White, T.A.; Sepe, A.; Johnson, K.O.; Stout, M.B.; Giorgadze, N.; et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife 2015, 4, e12997. [Google Scholar] [CrossRef]
- Lin, J.-R.; Shen, W.-L.; Yan, C.; Gao, P.-J. Downregulation of dynamin-related protein 1 contributes to impaired autophagic flux and angiogenic function in senescent endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1413–1422. [Google Scholar] [CrossRef] [Green Version]
- Cortegiani, A.; Ingoglia, G.; Ippolito, M.; Giarratano, A.; Einav, S. A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19. J. Crit. Care 2020. [Google Scholar] [CrossRef]
- Molina, J.M.; Delaugerre, C.; Goff, J.L.; Mela-Lima, B.; Ponscarme, D.; Goldwirt, L.; de Castro, N. No Evidence of Rapid Antiviral Clearance or Clinical Benefit with the Combination of Hydroxychloroquine and Azithromycin in Patients with Severe COVID-19 Infection. Médecine Mal. Infect. 2020. [Google Scholar] [CrossRef]
- Ozsvari, B.; Nuttall, J.R.; Sotgia, F.; Lisanti, M.P. Azithromycin and Roxithromycin define a new family of “senolytic” drugs that target senescent human fibroblasts. Aging (Albany. NY). 2018, 10, 3294–3307. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zu, S.; Deng, Y.Q.; Li, D.; Parvatiyar, K.; Quanquin, N.; Shang, J.; Sun, N.; Su, J.; Liu, Z.; et al. Azithromycin protects against Zika virus infection by upregulating virus-induced type I and III interferon responses. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.H.; Sugamata, R.; Hirose, T.; Suzuki, S.; Noguchi, Y.; Sugawara, A.; Ito, F.; Yamamoto, T.; Kawachi, S.; Akagawa, K.S.; et al. Azithromycin, a 15-membered macrolide antibiotic, inhibits influenza A(H1N1)pdm09 virus infection by interfering with virus internalization process. J. Antibiot. (Tokyo). 2019, 72, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Principi, N.; Blasi, F.; Esposito, S. Azithromycin use in patients with cystic fibrosis. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 1071–1079. [Google Scholar] [CrossRef]
- Menzel, M.; Akbarshahi, H.; Bjermer, L.; Uller, L. Azithromycin induces anti-viral effects in cultured bronchial epithelial cells from COPD patients. Sci. Rep. 2016, 6, 28698. [Google Scholar] [CrossRef] [Green Version]
- Bezzerri, V.; Piacenza, F.; Caporelli, N.; Malavolta, M.; Provinciali, M.; Cipolli, M. Is cellular senescence involved in cystic fibrosis? Respir. Res. 2019, 20, 32. [Google Scholar] [CrossRef]
- Kumar, M.; Seeger, W.; Voswinckel, R. Senescence-associated secretory phenotype and its possible role in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2014. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, Z.; Li, J.-W.; Zhao, H.; Wang, G.-Q. The cytokine release syndrome (CRS) of severe COVID-19 and Interleukin-6 receptor (IL-6R) antagonist Tocilizumab may be the key to reduce the mortality. Int. J. Antimicrob. Agents 2020, 105954. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [Green Version]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Kawamoto, S.; Ohtani, N.; Hara, E. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 2017, 108, 563–569. [Google Scholar] [CrossRef] [Green Version]
- Saleh, T.; Tyutynuk-Massey, L.; Cudjoe, E.K.; Idowu, M.O.; Landry, J.W.; Gewirtz, D.A. Non-cell autonomous effects of the senescence-associated secretory phenotype in cancer therapy. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojko, A.; Czarnecka-Herok, J.; Charzynska, A.; Dabrowski, M.; Sikora, E. Diversity of the Senescence Phenotype of Cancer Cells Treated with Chemotherapeutic Agents. Cells 2019, 8, 1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, D.G.A.; Stolzing, A. Cellular senescence: Immunosurveillance and future immunotherapy. Ageing Res. Rev. 2018, 43, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008. [Google Scholar] [CrossRef] [Green Version]
- Stokes, K.L.; Cortez-Retamozo, V.; Acosta, J.; Lauderback, B.; Robles-Oteiza, C.; Cicchini, M.; Pittet, M.J.; Feldser, D.M. Natural killer cells limit the clearance of senescent lung adenocarcinoma cells. Oncogenesis 2019, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Idda, M.L.; McClusky, W.G.; Lodde, V.; Munk, R.; Abdelmohsen, K.; Rossi, M.; Gorospe, M. Survey of senescent-cell markers with age in human tissues. Aging (Albany. NY) 2020, 12, 4052–4066. [Google Scholar] [CrossRef]
- Campisi, J. Cellular senescence and lung function during aging: Yin and Yang. Ann. Am. Thorac. Soc. 2016, 13, S402–S406. [Google Scholar] [CrossRef] [Green Version]
- Rashid, K.; Sundar, I.K.; Gerloff, J.; Li, D.; Rahman, I. Lung cellular senescence is independent of aging in a mouse model of COPD/emphysema. Sci. Rep. 2018, 8, 9023. [Google Scholar] [CrossRef] [Green Version]
- Perez-Lanzon, M.; Zitvogel, L.; Kroemer, G. Failure of immunosurveillance accelerates aging. Oncoimmunology 2019, 8, e1575117. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Zhao, J.; Bukata, C.; Wade, E.A.; McGowan, S.J.; Angelini, L.A.; Bank, M.P.; Gurkar, A.U.; McGuckian, C.A.; Calubag, M.F.; et al. Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Salmonowicz, H.; Gladyshev, V.N. Integrating cellular senescence with the concept of damage accumulation in aging: Relevance for clearance of senescent cells. Aging Cell 2019, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takasugi, M. Emerging roles of extracellular vesicles in cellular senescence and aging. Aging Cell 2018, 17, e12734. [Google Scholar] [CrossRef]
- Alibhai, F.J.; Lim, F.; Yeganeh, A.; DiStefano, P.V.; Binesh-Marvasti, T.; Belfiore, A.; Wlodarek, L.; Gustafson, D.; Millar, S.; Li, S.-H.; et al. Cellular senescence contributes to age-dependent changes in circulating extracellular vesicle cargo and function. Aging Cell 2020, e13103. [Google Scholar] [CrossRef] [Green Version]
- Jeon, O.H.; Wilson, D.R.; Clement, C.C.; Rathod, S.; Cherry, C.; Powell, B.; Lee, Z.; Khalil, A.M.; Green, J.J.; Campisi, J.; et al. Senescence cell–associated extracellular vesicles serve as osteoarthritis disease and therapeutic markers. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Mensà, E.; Guescini, M.; Giuliani, A.; Bacalini, M.G.; Ramini, D.; Corleone, G.; Ferracin, M.; Fulgenzi, G.; Graciotti, L.; Prattichizzo, F.; et al. Small extracellular vesicles deliver miR-21 and miR-217 as pro-senescence effectors to endothelial cells. J. Extracell. Vesicles 2020, 9, 1725285. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Crenshaw, B.J.; Gu, L.; Sims, B.; Matthews, Q.L. Exosome Biogenesis and Biological Function in Response to Viral Infections. Open Virol. J. 2018, 12, 134–148. [Google Scholar] [CrossRef] [Green Version]
- Reddel, R.R. Senescence: An antiviral defense that is tumor suppressive? Carcinogenesis 2010, 31, 19–26. [Google Scholar] [CrossRef]
- Moiseeva, O.; Mallette, F.A.; Mukhopadhyay, U.K.; Moores, A.; Ferbeyre, G. DNA damage signaling and p53-dependent senescence after prolonged beta-interferon stimulation. Mol. Biol. Cell 2006, 17, 1583–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, G.; Kucheryavenko, O.; Wordsworth, J.; von Zglinicki, T. The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech. Ageing Dev. 2018, 170, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.W.; Chan, F.K.M. Staying alive: Cell death in antiviral immunity. Mol. Cell 2014, 54, 273–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baz-Martínez, M.; Da Silva-Álvarez, S.; Rodríguez, E.; Guerra, J.; El Motiam, A.; Vidal, A.; Garciá-Caballero, T.; González-Barcia, M.; Sánchez, L.; Munõz-Fontela, C.; et al. Cell senescence is an antiviral defense mechanism. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Spyridopoulos, I.; Martin-Ruiz, C.; Hilkens, C.; Yadegarfar, M.E.; Isaacs, J.; Jagger, C.; Kirkwood, T.; von Zglinicki, T. CMV seropositivity and T-cell senescence predict increased cardiovascular mortality in octogenarians: Results from the Newcastle 85+ study. Aging Cell 2016, 15, 389–392. [Google Scholar] [CrossRef] [Green Version]
- Giacconi, R.; Maggi, F.; Macera, L.; Pistello, M.; Provinciali, M.; Giannecchini, S.; Martelli, F.; Spezia, P.G.; Mariani, E.; Galeazzi, R.; et al. Torquetenovirus (TTV) load is associated with mortality in Italian elderly subjects. Exp. Gerontol. 2018, 112, 103–111. [Google Scholar] [CrossRef]
- Giacconi, R.; Maggi, F.; Macera, L.; Spezia, P.G.; Pistello, M.; Provinciali, M.; Piacenza, F.; Basso, A.; Bürkle, A.; Moreno-Villanueva, M.; et al. Prevalence and loads of torquetenovirus (TTV) in the European MARK-AGE Study population. J. Gerontol. A. Biol. Sci. Med. Sci. 2019. [Google Scholar] [CrossRef]
- Maggi, F.; Focosi, D.; Statzu, M.; Bianco, G.; Costa, C.; Macera, L.; Spezia, P.G.; Medici, C.; Albert, E.; Navarro, D.; et al. Early Post-Transplant Torquetenovirus Viremia Predicts Cytomegalovirus Reactivations In Solid Organ Transplant Recipients. Sci. Rep. 2018, 8, 15490. [Google Scholar] [CrossRef]
- Heath, J.J.; Grant, M.D. The Immune Response Against Human Cytomegalovirus Links Cellular to Systemic Senescence. Cells 2020, 9, 766. [Google Scholar] [CrossRef] [Green Version]
- McElhaney, J.E.; Garneau, H.; Camous, X.; Dupuis, G.; Pawelec, G.; Baehl, S.; Tessier, D.; Frost, E.H.; Frasca, D.; Larbi, A.; et al. Predictors of the antibody response to influenza vaccination in older adults with type 2 diabetes. BMJ Open Diabetes Res. Care 2015, 3, e000140. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.C.; Partridge, A.T.; Tuzer, F.; Cohen, J.; Nacarelli, T.; Navas-Martín, S.; Sell, C.; Torres, C.; Martín-García, J. Induction of a senescence-like phenotype in cultured human fetal microglia during HIV-1 infection. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2018, 73, 1187–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafez, A.Y.; Luftig, M.A. Characterization of the EBV-induced persistent DNA damage response. Viruses 2017, 9. [Google Scholar]
- Chuprin, A.; Gal, H.; Biron-Shental, T.; Biran, A.; Amiel, A.; Rozenblatt, S.; Krizhanovsky, V. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev. 2013, 27, 2356–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abubakar, S.; Shu, M.H.; Johari, J.; Wong, P.F. Senescence affects endothelial cells susceptibility to dengue virus infection. Int. J. Med. Sci. 2014, 11, 538–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Du, Y.; Zheng, H.; Wang, G.; Li, R.; Chen, J.; Li, K. NS1 of H7N9 Influenza A Virus Induces NO-Mediated Cellular Senescence in Neuro2a Cells. Cell. Physiol. Biochem. 2017, 43, 1369–1380. [Google Scholar] [CrossRef] [PubMed]
- Bryan, T.M.; Reddel, R.R. SV40-induced immortalization of human cells. Crit. Rev. Oncog. 1994, 5, 331–357. [Google Scholar] [CrossRef] [PubMed]
- DiPaolo, J.A.; Popescu, N.C.; Alvarez, L.; Woodworth, C.D. Cellular and molecular alterations in human epithelial cells transformed by recombinant human papillomavirus DNA. Crit. Rev. Oncog. 1993, 4, 337–360. [Google Scholar]
- Kim, J.A.; Seong, R.K.; Shin, O.S. Enhanced viral replication by cellular replicative senescence. Immune Netw. 2016, 16, 286–295. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, H.; Yamamoto, K.; Nozato, S.; Takeda, M.; Fukada, S.; Inagaki, T.; Tsuchimochi, H.; Shirai, M.; Nozato, Y.; Fujimoto, T.; et al. Angiotensin-converting enzyme 2 deficiency accelerates and angiotensin 1-7 restores age-related muscle weakness in mice. J. Cachexia. Sarcopenia Muscle 2018, 9, 975–986. [Google Scholar] [CrossRef]
- Li, W.; Wang, W.; Li, Y.; Wang, W.; Wang, T.; Li, L.; Han, Z.; Wang, S.; Ma, D.; Wang, H. Proteomics analysis of normal and senescent NG108-15 cells: GRP78 plays a negative role in cisplatin-induced senescence in the NG108-15 cell line. PLoS ONE 2014, 9, e90114. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.; Guan, W.; Chen, R.; Wang, W.; Li, J.; Xu, K.; Li, C.; Ai, Q.; Lu, W.; Liang, H.; et al. Cancer patients in SARS-CoV-2 infection: A nationwide analysis in China. Lancet. Oncol. 2020, 21, 335–337. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagiv, A.; Krizhanovsky, V. Immunosurveillance of senescent cells: The bright side of the senescence program. Biogerontology 2013, 14, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elyahu, Y.; Hekselman, I.; Eizenberg-Magar, I.; Berner, O.; Strominger, I.; Schiller, M.; Mittal, K.; Nemirovsky, A.; Eremenko, E.; Vital, A.; et al. Aging promotes reorganization of the CD4 T cell landscape toward extreme regulatory and effector phenotypes. Sci. Adv. 2019, 5, eaaw8330. [Google Scholar] [CrossRef] [Green Version]
- Prata, L.G.P.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef]
- Chen, J.; Lau, Y.F.; Lamirande, E.W.; Paddock, C.D.; Bartlett, J.H.; Zaki, S.R.; Subbarao, K. Cellular Immune Responses to Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) Infection in Senescent BALB/c Mice: CD4+ T Cells Are Important in Control of SARS-CoV Infection. J. Virol. 2010, 84, 1289–1301. [Google Scholar] [CrossRef] [Green Version]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Grazioli, S.; Pugin, J. Mitochondrial damage-associated molecular patterns: From inflammatory signaling to human diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef]
- Ziegler, D.V.; Wiley, C.D.; Velarde, M.C. Mitochondrial effectors of cellular senescence: Beyond the free radical theory of aging. Aging Cell 2015, 14, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [PubMed] [Green Version]
- Shi, C.-S.; Qi, H.-Y.; Boularan, C.; Huang, N.-N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-9b Suppresses Innate Immunity by Targeting Mitochondria and the MAVS/TRAF3/TRAF6 Signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [Green Version]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The Role of Dynamin-Related Protein 1, a Mediator of Mitochondrial Fission, in Apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef] [Green Version]
- Karbowski, M.; Lee, Y.J.; Gaume, B.; Jeong, S.Y.; Frank, S.; Nechushtan, A.; Santel, A.; Fuller, M.; Smith, C.L.; Youle, R.J. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J. Cell Biol. 2002, 159, 931–938. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Wu, S.; Ren, H.; Gu, J. Klotho suppresses RIG-I-mediated senescence-associated inflammation. Nat. Cell Biol. 2011, 13, 254–262. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Hill, T.; Li, K.; Peters, C.J.; Tseng, C.-T.K. Severe Acute Respiratory Syndrome (SARS) Coronavirus-Induced Lung Epithelial Cytokines Exacerbate SARS Pathogenesis by Modulating Intrinsic Functions of Monocyte-Derived Macrophages and Dendritic Cells. J. Virol. 2009, 83, 3039–3048. [Google Scholar] [CrossRef] [Green Version]
- Barbier, V.; Lang, D.; Valois, S.; Rothman, A.L.; Medin, C.L. Dengue virus induces mitochondrial elongation through impairment of Drp1-triggered mitochondrial fission. Virology 2017, 500, 149–160. [Google Scholar] [CrossRef]
- Myrianthopoulos, V.; Evangelou, K.; Vasileiou, P.V.S.; Cooks, T.; Vassilakopoulos, T.P.; Pangalis, G.A.; Kouloukoussa, M.; Kittas, C.; Georgakilas, A.G.; Gorgoulis, V.G. Senescence and senotherapeutics: A new field in cancer therapy. Pharmacol. Ther. 2019, 193, 31–49. [Google Scholar] [CrossRef]
- Myrianthopoulos, V. The emerging field of senotherapeutic drugs. Future Med. Chem. 2018, 10, 2369–2372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malavolta, M.; Costarelli, L.; Giacconi, R.; Piacenza, F.; Basso, A.; Pierpaoli, E.; Marchegiani, F.; Cardelli, M.; Provinciali, M.; Mocchegiani, E. Modulators of cellular senescence: Mechanisms, promises, and challenges from in vitro studies with dietary bioactive compounds. Nutr. Res. 2014, 34, 1017–1035. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Noh, J.H.; Bodogai, M.; Martindale, J.L.; Yang, X.; Indig, F.E.; Basu, S.K.; Ohnuma, K.; Morimoto, C.; Johnson, P.F.; et al. Identification of senescent cell surface targetable protein DPP4. Genes Dev. 2017, 31, 1529–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubackova, S.; Davidova, E.; Rohlenova, K.; Stursa, J.; Werner, L.; Andera, L.; Dong, L.F.; Terp, M.G.; Hodny, Z.; Ditzel, H.J.; et al. Selective elimination of senescent cells by mitochondrial targeting is regulated by ANT2. Cell Death Differ. 2019, 26, 276–290. [Google Scholar] [CrossRef] [Green Version]
- Dörr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Däbritz, J.H.M.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef]
- Mizunoe, S.; Kadota, J.I.; Tokimatsu, I.; Kishi, K.; Nagai, H.; Nasu, M. Clarithromycin and azithromycin induce apoptosis of activated lymphocytes via down-regulation of Bcl-xL. Int. Immunopharmacol. 2004, 4, 1201–1207. [Google Scholar] [CrossRef]
- Orzalli, M.H.; Kagan, J.C. Apoptosis and Necroptosis as Host Defense Strategies to Prevent Viral Infection. Trends Cell Biol. 2017, 27, 800–809. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Espín, D.; Rovira, M.; Galiana, I.; Giménez, C.; Lozano-Torres, B.; Paez-Ribes, M.; Llanos, S.; Chaib, S.; Muñoz-Martín, M.; Ucero, A.C.; et al. A versatile drug delivery system targeting senescent cells. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Agent | Study Related to COVID-19 Therapy | Target/Pathways Related to Cellular Senescence | References |
|---|---|---|---|
| Tocilizumab (atlizumab) | Clinical trial (ChiCTR2000029765) | Blocker of IL-6R, IL-6 is among the most common SASP factor | [28,29] |
| Hydroxychloroquine and azithromycin | Clinical Trial (NCT04322396) and other pilot studies | Azithromycin exerts senolytic activity of human senescent lung fibroblasts; Hydroxychloroquine induces senescence in endothelial cells | [18,20,21] |
| Ruxolitinib | Bioinformatic study (supported by BenevolentAI) Clinical trial (NCT04331665) | JAK inhibitor, SASP suppressor | [15,16,17] |
| Rapamycin | Network-based drug repurposing methodology for HCoV | Inhibitor of mTOR, SASP suppressor | [12,13] |
| Melatonin | Network-based drug repurposing methodology for HCoV | SASP suppressor in lung fibroblasts | [12,14] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malavolta, M.; Giacconi, R.; Brunetti, D.; Provinciali, M.; Maggi, F. Exploring the Relevance of Senotherapeutics for the Current SARS-CoV-2 Emergency and Similar Future Global Health Threats. Cells 2020, 9, 909. https://doi.org/10.3390/cells9040909
Malavolta M, Giacconi R, Brunetti D, Provinciali M, Maggi F. Exploring the Relevance of Senotherapeutics for the Current SARS-CoV-2 Emergency and Similar Future Global Health Threats. Cells. 2020; 9(4):909. https://doi.org/10.3390/cells9040909
Chicago/Turabian StyleMalavolta, Marco, Robertina Giacconi, Dario Brunetti, Mauro Provinciali, and Fabrizio Maggi. 2020. "Exploring the Relevance of Senotherapeutics for the Current SARS-CoV-2 Emergency and Similar Future Global Health Threats" Cells 9, no. 4: 909. https://doi.org/10.3390/cells9040909