Adenosine A2A Receptor Antagonists Affects NMDA Glutamate Receptor Function. Potential to Address Neurodegeneration in Alzheimer’s Disease

 ,
,  and
and
Abstract
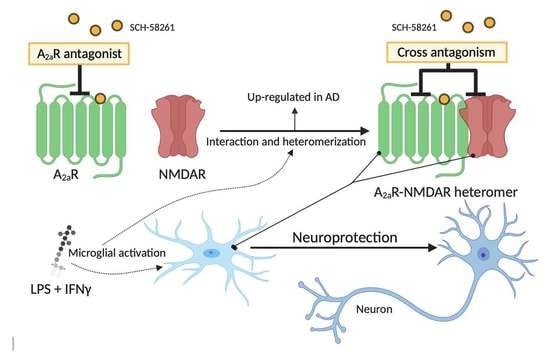
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Fusion Proteins
2.3. APP Transgenic Mouse Model of Alzheimer’s Disease (AD)
2.4. Cell Culture and Transfection
2.5. Immunocytochemistry
2.6. Bioluminescence Resonance Energy Transfer (BRET) Assays
2.7. Cyclic Adenylic Acid (cAMP) Determination
2.8. Extracellular Signal-Regulated Kinase (ERK) Phosphorylation Determination
2.9. Assessment of Dynamic Mass Redistribution (DMR)
2.10. Determination of Cytoplasmic Calcium Ion Level Increase
2.11. In Situ Proximity Ligation Assay (PLA)
2.12. Statistical Analysis
3. Results
3.1. NMDA Receptors May Directly Interact with Adenosine A2A Receptors
3.2. Functional Properties of A2A-NMDA Receptor Heteromer Complex
3.3. A2A-NMDA Receptor Heteromer Complex Expression in Resting and Activated Microglia
3.4. A2AR-NMDAR Heteromer Expression is Elevated in Primary Microglia from APPSw/Ind Mice
3.5. A2AR Activation Negatively Modulates NMDA Receptor Signaling in Microglia but it Increases NMDAR Signaling in Neurons from APPSw,Ind Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lipton, S. Pathologically-Activated Therapeutics for Neuroprotection: Mechanism of NMDA Receptor Block by Memantine and S-Nitrosylation. Curr. Drug Targets 2007, 8, 261–632. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat. Rev. Drug Discov. 2006, 5, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Parsons, C.G.; Stöffler, A.; Danysz, W. Memantine: A NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system-too little activation is bad, too much is even worse. Neuropharmacology 2007, 53, 699–723. [Google Scholar] [CrossRef]
- Kisby, B.; Jarrell, J.T.; Agar, M.E.; Cohen, D.S.; Rosin, E.R.; Cahill, C.M.; Rogers, J.T.; Huang, X. Alzheimer’s Disease and Its Potential Alternative Therapeutics. J. Alzheimer’s Dis. Park. 2019, 9, 477. [Google Scholar] [CrossRef]
- Rosin, D.L.; Robeva, A.; Woodard, R.L.; Guyenet, P.G.; Linden, J. Immunohistochemical Localization of Adenosine A 2A Receptors in the Rat Central Nervous System. J. Comp. Neurol. 1998, 401, 163–186. [Google Scholar] [CrossRef]
- Kondo, T.; Mizuno, Y. Japanese Istradefylline Study Group A long-term study of istradefylline safety and efficacy in patients with Parkinson disease. Clin. Neuropharmacol. 2015, 38, 41–46. [Google Scholar] [CrossRef]
- Saki, M.; Yamada, K.; Koshimura, E.; Sasaki, K.; Kanda, T. In vitro pharmacological profile of the A2A receptor antagonist istradefylline. Naunyn. Schmiedebergs. Arch. Pharmacol. 2013, 386, 963–972. [Google Scholar] [CrossRef]
- Mizuno, Y.; Kondo, T. Adenosine A2A receptor antagonist istradefylline reduces daily OFF time in Parkinson’s disease. Mov. Disord. 2013, 28, 1138–1141. [Google Scholar] [CrossRef] [Green Version]
- Schubert, P.; Kreutzberg, G.W. Cerebral protection by adenosine. Acta Neurochir. Suppl. 1993, 57, 80–88. [Google Scholar]
- Franco, R.; Reyes-Resina, I.; Aguinaga, D.; Lillo, A.; Jiménez, J.; Raïch, I.; Borroto-Escuela, D.O.; Ferreiro-Vera, C.; Canela, E.I.; Sánchez de Medina, V.; et al. Potentiation of cannabinoid signaling in microglia by adenosine A2A receptor antagonists. Glia 2019, 67, 2410–2423. [Google Scholar] [CrossRef]
- Gussago, C.; Arosio, B.; Casati, M.; Ferri, E.; Gualandris, F.; Tedone, E.; Nicolini, P.; Rossi, P.D.; Abbate, C.; Mari, D. Different adenosine A2A receptor expression in peripheral cells from elderly patients with vascular dementia and Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 40, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Burnouf, S.; Ferry, B.; Batalha, V.L.; Coelho, J.E.; Baqi, Y.; Malik, E.; Mariciniak, E.; Parrot, S.; Van Der Jeugd, A.; et al. A2A adenosine receptor deletion is protective in a mouse model of Tauopathy. Mol. Psychiatry 2016, 21, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faivre, E.; Coelho, J.E.; Zornbach, K.; Malik, E.; Baqi, Y.; Schneider, M.; Cellai, L.; Carvalho, K.; Sebda, S.; Figeac, M.; et al. Beneficial Effect of a Selective Adenosine A2A Receptor Antagonist in the APPswe/PS1dE9 Mouse Model of Alzheimer’s Disease. Front. Mol. Neurosci. 2018, 11, 235. [Google Scholar] [CrossRef] [PubMed]
- Temido-Ferreira, M.; Ferreira, D.G.; Batalha, V.L.; Marques-Morgado, I.; Coelho, J.E.; Pereira, P.; Gomes, R.; Pinto, A.; Carvalho, S.; Canas, P.M.; et al. Age-related shift in LTD is dependent on neuronal adenosine A2A receptors interplay with mGluR5 and NMDA receptors. Mol. Psychiatry 2018, 1–25. [Google Scholar] [CrossRef]
- Silva, A.C.; Lemos, C.; Gonçalves, F.Q.; Pliássova, A.V.; Machado, N.J.; Silva, H.B.; Canas, P.M.; Cunha, R.A.; Lopes, J.P.; Agostinho, P. Blockade of adenosine A2A receptors recovers early deficits of memory and plasticity in the triple transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 2018, 117, 72–81. [Google Scholar] [CrossRef]
- Streit, W.J.; Kreutzberg, G.W. Lectin binding by resting and reactive microglia. J. Neurocytol. 1987, 16, 249–260. [Google Scholar] [CrossRef]
- Streit, W.J. Microglia and neuroprotection: Implications for Alzheimer’s disease. Brain Res. Brain Res. Rev. 2005, 48, 234–239. [Google Scholar] [CrossRef]
- Wendt, S.; Wogram, E.; Korvers, L.; Kettenmann, H. Experimental cortical spreading depression induces NMDA receptor dependent potassium currents in microglia. J. Neurosci. 2016, 36, 6165–6174. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Zhao, Y.; Chen, X.; Zhu, M.; Miao, C. Propofol attenuates BV2 microglia inflammation via NMDA receptor inhibition. Can. J. Physiol. Pharmacol. 2018, 96, 241–248. [Google Scholar] [CrossRef] [Green Version]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Pinilla, E.; Rodríguez-Pérez, A.I.I.; Navarro, G.; Aguinaga, D.; Moreno, E.; Lanciego, J.L.L.; Labandeira-García, J.L.L.; Franco, R. Dopamine D2 and angiotensin II type 1 receptors form functional heteromers in rat striatum. Biochem. Pharmacol. 2015, 96, 131–142. [Google Scholar] [CrossRef]
- Newell, E.; Exo, J.; Verrier, J.; Jackson, T.; Gillespie, D.; Janesko-Feldman, K.; Kochanek, P.; Jackson, E. 2′,3′-cAMP, 3′-AMP, 2′-AMP and adenosine inhibit TNF-α and CXCL10 production from activated primary murine microglia via A2A receptors. Brain Res. 2015, 1594, 27–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hradsky, J.; Mikhaylova, M.; Karpova, A.; Kreutz, M.R.; Zuschratter, W. Super-resolution microscopy of the neuronal calcium-binding proteins Calneuron-1 and Caldendrin. Methods Mol. Biol. 2013, 963, 147–169. [Google Scholar] [PubMed]
- Chen, T.-W.; Wardill, T.J.; Sun, Y.; Pulver, S.R.; Renninger, S.L.; Baohan, A.; Schreiter, E.R.; Kerr, R.A.; Orger, M.B.; Jayaraman, V.; et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013, 499, 295–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferré, S.; Baler, R.; Bouvier, M.; Caron, M.G.; Devi, L.A.; Durroux, T.; Fuxe, K.; George, S.R.; Javitch, J.A.; Lohse, M.J.; et al. Building a new conceptual framework for receptor heteromers. Nat. Chem. Biol. 2009, 5, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Franco, R.; Martínez-Pinilla, E.; Lanciego, J.L.; Navarro, G. Basic Pharmacological and Structural Evidence for Class A G-Protein-Coupled Receptor Heteromerization. Front. Pharmacol. 2016, 7, 76. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Kodis, E.J.; Choi, S.; Swanson, E.; Ferreira, G.; Bloom, G.S. N-methyl-D-aspartate receptor–mediated calcium influx connects amyloid-β oligomers to ectopic neuronal cell cycle reentry in Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 1302–1312. [Google Scholar] [CrossRef]
- Stockwell, J.; Jakova, E.; Cayabyab, F.S. Adenosine A1 and A2A Receptors in the Brain: Current Research and Their Role in Neurodegeneration. Molecules 2017, 22, 676. [Google Scholar] [CrossRef] [Green Version]
- Constantino, L.C.; Pamplona, F.A.; Matheus, F.C.; Ludka, F.K.; Gomez-Soler, M.; Ciruela, F.; Boeck, C.R.; Prediger, R.D.; Tasca, C.I. Adenosine A1 receptor activation modulates N-methyl-d-aspartate (NMDA) preconditioning phenotype in the brain. Behav. Brain Res. 2015, 282, 103–110. [Google Scholar] [CrossRef]
- Gui, L.; Duan, W.; Tian, H.; Li, C.; Zhu, J.; Chen, J.F.; Zheng, J. Adenosine A2Areceptor deficiency reduces striatal glutamate outflow and attenuates brain injury induced by transient focal cerebral ischemia in mice. Brain Res. 2009, 1297, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Schwarzschild, M.A.; Chen, J.-F.; Ascherio, A. Caffeinated clues and the promise of adenosine A(2A) antagonists in PD. Neurology 2002, 58, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-F. Adenosine receptor control of cognition in normal and disease. Int. Rev. Neurobiol. 2014, 119, 257–307. [Google Scholar] [PubMed]
- Chen, J.-F.; Sonsalla, P.K.; Pedata, F.; Melani, A.; Domenici, M.R.; Popoli, P.; Geiger, J.; Lopes, L.V.; de Mendonça, A. Adenosine A2A receptors and brain injury: Broad spectrum of neuroprotection, multifaceted actions and “fine tuning” modulation. Prog. Neurobiol. 2007, 83, 310–331. [Google Scholar] [CrossRef]
- Fuxe, K.; Ferré, S.; Canals, M.; Torvinen, M.; Terasmaa, A.; Marcellino, D.; Goldberg, S.R.; Staines, W.; Jacobsen, K.X.; Lluis, C.; et al. Adenosine A2A and Dopamine D2 Heteromeric Receptor Complexes and Their Function. J. Mol. Neurosci. 2005, 26, 209–220. [Google Scholar] [CrossRef]
- Fuxe, K.; Ferré, S.; Genedani, S.; Franco, R.; Agnati, L.F. Adenosine receptor-dopamine receptor interactions in the basal ganglia and their relevance for brain function. Physiol. Behav. 2007, 92, 210–217. [Google Scholar] [CrossRef]
- Da Silva, S.V.; Haberl, M.G.; Zhang, P.; Bethge, P.; Lemos, C.; Gonçalves, N.; Gorlewicz, A.; Malezieux, M.; Gonçalves, F.Q.; Grosjean, N.; et al. Early synaptic deficits in the APP/PS1 mouse model of Alzheimer’s disease involve neuronal adenosine A2A receptors. Nat. Commun. 2016, 7, 11915. [Google Scholar] [CrossRef]
- Mouro, F.M.; Rombo, D.M.; Dias, R.B.; Ribeiro, J.A.; Sebastião, A.M. Adenosine A2A receptors facilitate synaptic NMDA currents in CA1 pyramidal neurons. Br. J. Pharmacol. 2018, 175, 4386–4397. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, F.Q.; Lopes, J.P.; Silva, H.B.; Lemos, C.; Silva, A.C.; Gonçalves, N.; Tomé, Â.R.; Ferreira, S.G.; Canas, P.M.; Rial, D.; et al. Synaptic and memory dysfunction in a β-amyloid model of early Alzheimer’s disease depends on increased formation of ATP-derived extracellular adenosine. Neurobiol. Dis. 2019, 132. [Google Scholar] [CrossRef]
- Carvalho, K.; Faivre, E.; Pietrowski, M.J.; Marques, X.; Gomez-Murcia, V.; Deleau, A.; Huin, V.; Hansen, J.N.; Kozlov, S.; Danis, C.; et al. Exacerbation of C1q dysregulation, synaptic loss and memory deficits in tau pathology linked to neuronal adenosine A2A receptor. Brain 2019, 142, 3636–3654. [Google Scholar] [CrossRef]
- Angulo, E.; Casadó, V.; Mallol, J.; Canela, E.I.; Viñals, F.; Ferrer, I.; Lluis, C.; Franco, R. A1 adenosine receptors accumulate in neurodegenerative structures in Alzheimer disease and mediate both amyloid precursor protein processing and tau phosphorylation and translocation. Brain Pathol. 2003, 13, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Catafau, A.; Bullich, S. Non-Amyloid PET Imaging Biomarkers for Neurodegeneration: Focus on Tau, Alpha-Synuclein and Neuroinflammation. Curr. Alzheimer Res. 2016, 14, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Saura, J.; Angulo, E.; Ejarque, A.; Casado, V.; Tusell, J.M.; Moratalla, R.; Chen, J.-F.F.; Schwarzschild, M.A.; Lluis, C.; Franco, R.; et al. Adenosine A2A receptor stimulation potentiates nitric oxide release by activated microglia. J. Neurochem. 2005, 95, 919–929. [Google Scholar] [CrossRef]
- Rebola, N.; Simões, A.P.; Canas, P.M.; Tomé, A.R.; Andrade, G.M.; Barry, C.E.; Agostinho, P.M.; Lynch, M.A.; Cunha, R.A. Adenosine A2A receptors control neuroinflammation and consequent hippocampal neuronal dysfunction. J. Neurochem. 2011, 117, 100–111. [Google Scholar] [CrossRef]
- Cunha, R.A. How Does Adenosine Control Neuronal Dysfunction and Neurodegeneration? Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2016; Volume 139, pp. 1019–1055. [Google Scholar]
- Santiago, A.R.; Baptista, F.I.; Santos, P.F.; Cristóvão, G.; Ambrósio, A.F.; Cunha, R.A.; Gomes, C.A.; Pintor, J. Role of microglia adenosine A2A receptors in retinal and brain neurodegenerative diseases. Mediat. Inflamm. 2014, 2014, 465694. [Google Scholar] [CrossRef]
- Eskelinen, M.H.; Kivipelto, M. Caffeine as a protective factor in dementia and Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, S167–S174. [Google Scholar] [CrossRef] [Green Version]
- Franco, R.; Aguinaga, D.; Jiménez, J.; Lillo, J.; Martínez-Pinilla, E.; Navarro, G. Biased receptor functionality versus biased agonism in G-protein-coupled receptors. Biomol. Concepts 2018, 9, 143–154. [Google Scholar] [CrossRef]
- Navarro, G.; Borroto-Escuela, D.; Angelats, E.; Etayo, I.; Reyes-Resina, I.; Pulido-Salgado, M.; Rodríguez-Pérez, A.; Canela, E.; Saura, J.; Lanciego, J.L.; et al. Receptor-heteromer mediated regulation of endocannabinoid signaling in activated microglia. Role of CB1 and CB2 receptors and relevance for Alzheimer’s disease and levodopa-induced dyskinesia. Brain. Behav. Immun. 2018, 67, 139–151. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P. Chronic glutamate toxicity in neurodegenerative diseases-What is the evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef]
- Lopes, L.V.; Sebastião, A.M.; Ribeiro, J.A. Adenosine and related drugs in brain diseases: Present and future in clinical trials. Curr. Top. Med. Chem. 2011, 11, 1087–1101. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco, R.; Rivas-Santisteban, R.; Casanovas, M.; Lillo, A.; Saura, C.A.; Navarro, G. Adenosine A2A Receptor Antagonists Affects NMDA Glutamate Receptor Function. Potential to Address Neurodegeneration in Alzheimer’s Disease. Cells 2020, 9, 1075. https://doi.org/10.3390/cells9051075
Franco R, Rivas-Santisteban R, Casanovas M, Lillo A, Saura CA, Navarro G. Adenosine A2A Receptor Antagonists Affects NMDA Glutamate Receptor Function. Potential to Address Neurodegeneration in Alzheimer’s Disease. Cells. 2020; 9(5):1075. https://doi.org/10.3390/cells9051075
Chicago/Turabian StyleFranco, Rafael, Rafael Rivas-Santisteban, Mireia Casanovas, Alejandro Lillo, Carlos A. Saura, and Gemma Navarro. 2020. "Adenosine A2A Receptor Antagonists Affects NMDA Glutamate Receptor Function. Potential to Address Neurodegeneration in Alzheimer’s Disease" Cells 9, no. 5: 1075. https://doi.org/10.3390/cells9051075