Progerin Expression Induces Inflammation, Oxidative Stress and Senescence in Human Coronary Endothelial Cells

 , , , and
, , , and
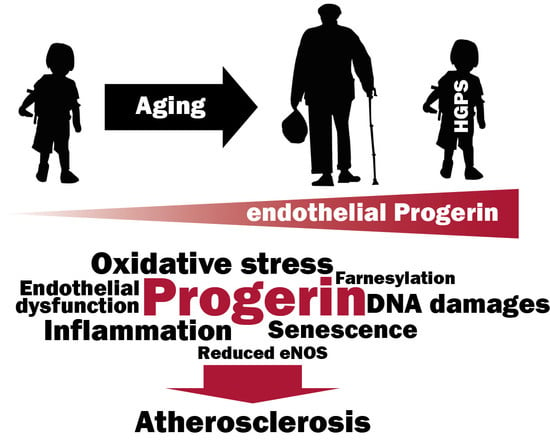
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Adenovirus Production and Adenoviral-Mediated Expression of WT-Prelamin A or Progerin in HCAECs
2.3. Progerin Detection and Nuclear Shape
2.4. Endothelial Dysfunction and Inflammation
2.5. Peripheral Blood Mononuclear Cell (PBMC) Adhesion Assay
2.6. Oxidative Stress Assay
2.7. Cellular Senescence Assay
2.8. Statistical Analysis
3. Results
3.1. Progerin Accumulates at the Nuclear Periphery and Disrupts the Nuclear Shape of Endothelial Cells
3.2. Progerin Expression Induces Endothelial Cell Inflammation and Reduces NO Synthase Expression
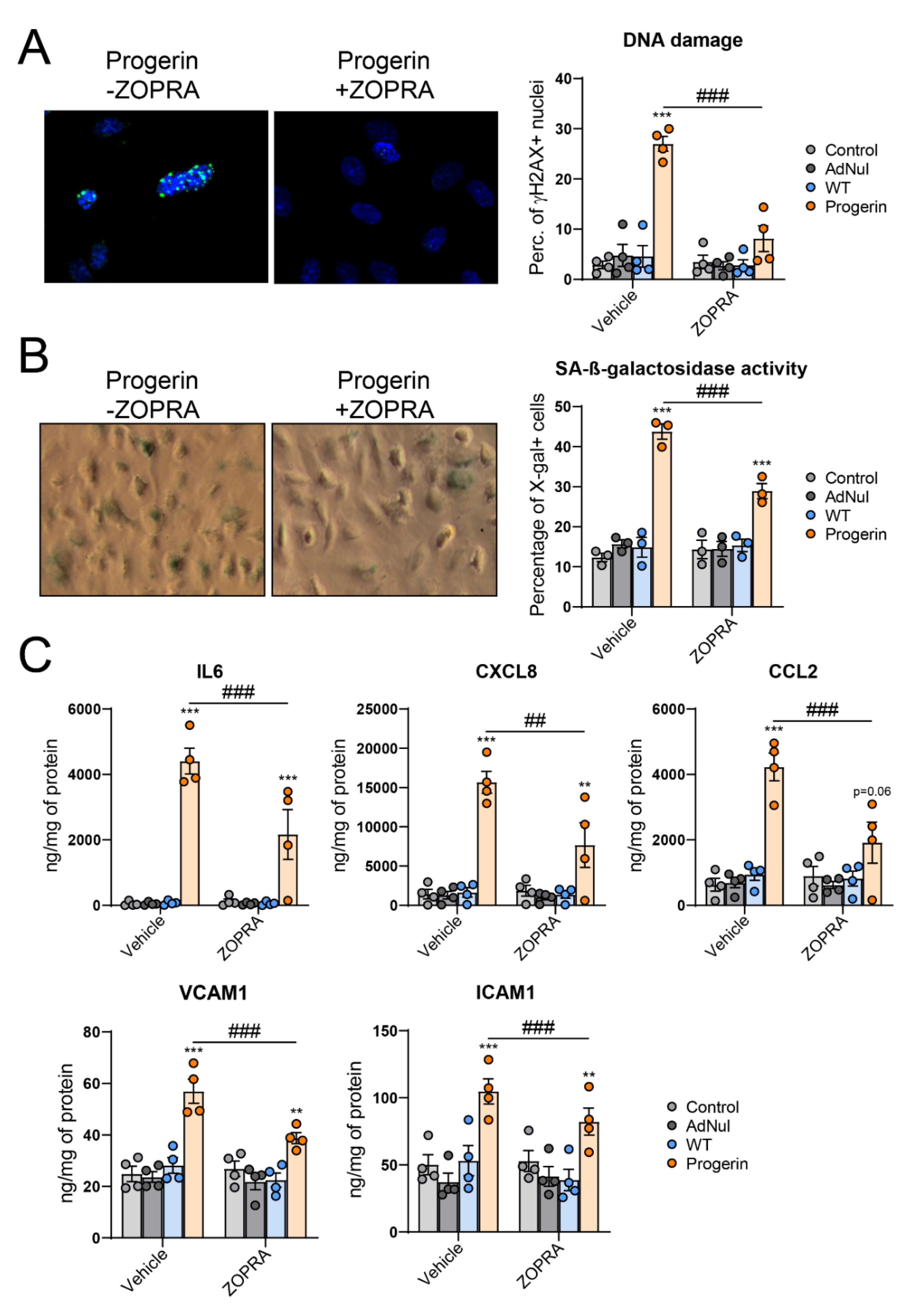
3.3. Endothelial Expression of Progerin Induces Oxidative Stress, DNA Damage and Senescence
3.4. Inhibition of Progerin Prenylation Partly Prevents Endothelial Defects
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin a truncation in Hutchinson–Gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of Hutchinson–Gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechat, T.; Adam, S.A.; Taimen, P.; Shimi, T.; Goldman, R.D. Nuclear lamins. Cold Spring Harb. Perspect. Biol. 2010, 2, a000547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrigan, D.P.; Kuszczak, D.; Rusinol, A.E.; Thewke, D.P.; Hrycyna, C.A.; Michaelis, S.; Sinensky, M.S. Prelamin A endoproteolytic processing in vitro by recombinant Zmpste24. Biochem. J. 2005, 387, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worman, H.J.; Michaelis, S. Permanently Farnesylated Prelamin A, Progeria, and Atherosclerosis. Circulation 2018, 138, 283–286. [Google Scholar] [CrossRef]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson–Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [Green Version]
- Stehbens, W.E.; Wakefield, S.J.; Gilbert-Barness, E.; Olson, R.E.; Ackerman, J. Histological and ultrastructural features of atherosclerosis in progeria. Cardiovasc. Pathol. 1999, 8, 29–39. [Google Scholar] [CrossRef]
- Olive, M.; Harten, I.; Mitchell, R.; Beers, J.K.; Djabali, K.; Cao, K.; Erdos, M.R.; Blair, C.; Funke, B.; Smoot, L.; et al. Cardiovascular pathology in Hutchinson–Gilford progeria: Correlation with the vascular pathology of aging. Arterioscler Thromb. Vasc. Biol. 2010, 30, 2301–2309. [Google Scholar] [CrossRef] [Green Version]
- Hamczyk, M.R.; Villa-Bellosta, R.; Gonzalo, P.; Andres-Manzano, M.J.; Nogales, P.; Bentzon, J.F.; Lopez-Otin, C.; Andres, V. Vascular Smooth Muscle-Specific Progerin Expression Accelerates Atherosclerosis and Death in a Mouse Model of Hutchinson–Gilford Progeria Syndrome. Circulation 2018, 138, 266–282. [Google Scholar] [CrossRef]
- Osorio, F.G.; Navarro, C.L.; Cadinanos, J.; Lopez-Mejia, I.C.; Quiros, P.M.; Bartoli, C.; Rivera, J.; Tazi, J.; Guzman, G.; Varela, I.; et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci. Transl. Med. 2011, 3, 106ra107. [Google Scholar] [CrossRef] [PubMed]
- Varga, R.; Eriksson, M.; Erdos, M.R.; Olive, M.; Harten, I.; Kolodgie, F.; Capell, B.C.; Cheng, J.; Faddah, D.; Perkins, S.; et al. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson–Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 3250–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClintock, D.; Gordon, L.B.; Djabali, K. Hutchinson–Gilford progeria mutant lamin A primarily targets human vascular cells as detected by an anti-Lamin A G608G antibody. Proc. Natl. Acad. Sci. USA 2006, 103, 2154–2159. [Google Scholar] [CrossRef] [Green Version]
- Osmanagic-Myers, S.; Kiss, A.; Manakanatas, C.; Hamza, O.; Sedlmayer, F.; Szabo, P.L.; Fischer, I.; Fichtinger, P.; Podesser, B.K.; Eriksson, M.; et al. Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse. J. Clin. Invest. 2019, 129, 531–545. [Google Scholar] [CrossRef] [Green Version]
- Del Campo, L.; Sanchez-Lopez, A.; Gonzalez-Gomez, C.; Andres-Manzano, M.J.; Dorado, B.; Andres, V. Vascular Smooth Muscle Cell-Specific Progerin Expression Provokes Contractile Impairment in a Mouse Model of Hutchinson–Gilford Progeria Syndrome that Is Ameliorated by Nitrite Treatment. Cells 2020, 9, 656. [Google Scholar] [CrossRef] [Green Version]
- Donato, A.J.; Morgan, R.G.; Walker, A.E.; Lesniewski, L.A. Cellular and molecular biology of aging endothelial cells. J. Mol. Cell Cardiol. 2015, 89, 122–135. [Google Scholar] [CrossRef] [Green Version]
- Barinda, A.J.; Ikeda, K.; Nugroho, D.B.; Wardhana, D.A.; Sasaki, N.; Honda, S.; Urata, R.; Matoba, S.; Hirata, K.I.; Emoto, N. Endothelial progeria induces adipose tissue senescence and impairs insulin sensitivity through senescence associated secretory phenotype. Nat. Commun. 2020, 11, 481. [Google Scholar] [CrossRef] [Green Version]
- Ashapkin, V.V.; Kutueva, L.I.; Kurchashova, S.Y.; Kireev, I.I. Are There Common Mechanisms Between the Hutchinson–Gilford Progeria Syndrome and Natural Aging? Front. Genet. 2019, 10, 455. [Google Scholar] [CrossRef] [Green Version]
- Bidault, G.; Garcia, M.; Vantyghem, M.C.; Ducluzeau, P.H.; Morichon, R.; Thiyagarajah, K.; Moritz, S.; Capeau, J.; Vigouroux, C.; Bereziat, V. Lipodystrophy-linked LMNA p.R482W mutation induces clinical early atherosclerosis and in vitro endothelial dysfunction. Arterioscler Thromb. Vasc. Biol. 2013, 33, 2162–2171. [Google Scholar] [CrossRef] [Green Version]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaupere, C.; Garcia, M.; Larghero, J.; Feve, B.; Capeau, J.; Lagathu, C. The HIV proteins Tat and Nef promote human bone marrow mesenchymal stem cell senescence and alter osteoblastic differentiation. Aging Cell 2015, 14, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Capell, B.C.; Erdos, M.R.; Madigan, J.P.; Fiordalisi, J.J.; Varga, R.; Conneely, K.N.; Gordon, L.B.; Der, C.J.; Cox, A.D.; Collins, F.S. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson–Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 12879–12884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Auclair, M.; Guenantin, A.C.; Fellahi, S.; Garcia, M.; Capeau, J. HIV antiretroviral drugs, dolutegravir, maraviroc and ritonavir-boosted atazanavir use different pathways to affect inflammation, senescence and insulin sensitivity in human coronary endothelial cells. PLoS ONE 2020, 15, e0226924. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Kubben, N.; Zhang, W.; Wang, L.; Voss, T.C.; Yang, J.; Qu, J.; Liu, G.H.; Misteli, T. Repression of the Antioxidant NRF2 Pathway in Premature Aging. Cell 2016, 165, 1361–1374. [Google Scholar] [CrossRef] [Green Version]
- Burtenshaw, D.; Kitching, M.; Redmond, E.M.; Megson, I.L.; Cahill, P.A. Reactive Oxygen Species (ROS), Intimal Thickening, and Subclinical Atherosclerotic Disease. Front. Cardiovasc. Med. 2019, 6, 89. [Google Scholar] [CrossRef]
- Van Nguyen, T.; Puebla-Osorio, N.; Pang, H.; Dujka, M.E.; Zhu, C. DNA damage-induced cellular senescence is sufficient to suppress tumorigenesis: A mouse model. J. Exp. Med. 2007, 204, 1453–1461. [Google Scholar] [CrossRef]
- Varela, I.; Pereira, S.; Ugalde, A.P.; Navarro, C.L.; Suarez, M.F.; Cau, P.; Cadinanos, J.; Osorio, F.G.; Foray, N.; Cobo, J.; et al. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat. Med. 2008, 14, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Harhouri, K.; Frankel, D.; Bartoli, C.; Roll, P.; De Sandre-Giovannoli, A.; Levy, N. An overview of treatment strategies for Hutchinson–Gilford Progeria syndrome. Nucleus 2018, 9, 246–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osorio, F.G.; Barcena, C.; Soria-Valles, C.; Ramsay, A.J.; de Carlos, F.; Cobo, J.; Fueyo, A.; Freije, J.M.; Lopez-Otin, C. Nuclear lamina defects cause ATM-dependent NF-kappaB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012, 26, 2311–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P. Inflammation in atherosclerosis. Arterioscler Thromb. Vasc. Biol. 2012, 32, 2045–2051. [Google Scholar] [CrossRef] [Green Version]
- Krabbe, K.S.; Pedersen, M.; Bruunsgaard, H. Inflammatory mediators in the elderly. Exp. Gerontol. 2004, 39, 687–699. [Google Scholar] [CrossRef]
- Donato, A.J.; Eskurza, I.; Silver, A.E.; Levy, A.S.; Pierce, G.L.; Gates, P.E.; Seals, D.R. Direct evidence of endothelial oxidative stress with aging in humans: Relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kappaB. Circ. Res. 2007, 100, 1659–1666. [Google Scholar] [CrossRef] [Green Version]
- Donato, A.J.; Black, A.D.; Jablonski, K.L.; Gano, L.B.; Seals, D.R. Aging is associated with greater nuclear NF kappa B, reduced I kappa B alpha, and increased expression of proinflammatory cytokines in vascular endothelial cells of healthy humans. Aging Cell 2008, 7, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Qin, W.; Tang, X.; Meng, Y.; Hu, W.; Zhang, S.; Qian, M.; Liu, Z.; Cao, X.; Pang, Q.; et al. Vascular endothelium-targeted Sirt7 gene therapy rejuvenates blood vessels and extends life span in a Hutchinson–Gilford progeria model. Sci. Adv. 2020, 6, eaay5556. [Google Scholar] [CrossRef] [Green Version]
- Lavi, S.; Yang, E.H.; Prasad, A.; Mathew, V.; Barsness, G.W.; Rihal, C.S.; Lerman, L.O.; Lerman, A. The interaction between coronary endothelial dysfunction, local oxidative stress, and endogenous nitric oxide in humans. Hypertension 2008, 51, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Pekovic, V.; Gibbs-Seymour, I.; Markiewicz, E.; Alzoghaibi, F.; Benham, A.M.; Edwards, R.; Wenhert, M.; von Zglinicki, T.; Hutchison, C.J. Conserved cysteine residues in the mammalian lamin A tail are essential for cellular responses to ROS generation. Aging Cell 2011, 10, 1067–1079. [Google Scholar] [CrossRef]
- Chen, B.; Lu, Y.; Chen, Y.; Cheng, J. The role of Nrf2 in oxidative stress-induced endothelial injuries. J. Endocrinol. 2015, 225, R83–R99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakkar, M.; Van der Heiden, K.; Luong, L.A.; Chaudhury, H.; Cuhlmann, S.; Hamdulay, S.S.; Krams, R.; Edirisinghe, I.; Rahman, I.; Carlsen, H.; et al. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arter. Thromb. Vasc. Biol. 2009, 29, 1851–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satta, S.; Mahmoud, A.M.; Wilkinson, F.L.; Yvonne Alexander, M.; White, S.J. The Role of Nrf2 in Cardiovascular Function and Disease. Oxid. Med. Cell Longev. 2017, 2017, 9237263. [Google Scholar] [CrossRef] [PubMed]
- Lefevre, C.; Auclair, M.; Boccara, F.; Bastard, J.P.; Capeau, J.; Vigouroux, C.; Caron-Debarle, M. Premature senescence of vascular cells is induced by HIV protease inhibitors: Implication of prelamin A and reversion by statin. Arter. Thromb. Vasc. Biol. 2010, 30, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glynn, M.W.; Glover, T.W. Incomplete processing of mutant lamin A in Hutchinson–Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum. Mol. Genet. 2005, 14, 2959–2969. [Google Scholar] [CrossRef]
- Toth, J.I.; Yang, S.H.; Qiao, X.; Beigneux, A.P.; Gelb, M.H.; Moulson, C.L.; Miner, J.H.; Young, S.G.; Fong, L.G. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc. Natl. Acad. Sci. USA 2005, 102, 12873–12878. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Bergo, M.O.; Toth, J.I.; Qiao, X.; Hu, Y.; Sandoval, S.; Meta, M.; Bendale, P.; Gelb, M.H.; Young, S.G.; et al. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson–Gilford progeria syndrome mutation. Proc. Natl. Acad. Sci. USA 2005, 102, 10291–10296. [Google Scholar] [CrossRef] [Green Version]
- Mallampalli, M.P.; Huyer, G.; Bendale, P.; Gelb, M.H.; Michaelis, S. Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson–Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 14416–14421. [Google Scholar] [CrossRef] [Green Version]
- Capell, B.C.; Olive, M.; Erdos, M.R.; Cao, K.; Faddah, D.A.; Tavarez, U.L.; Conneely, K.N.; Qu, X.; San, H.; Ganesh, S.K.; et al. A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a progeria mouse model. Proc. Natl. Acad. Sci. USA 2008, 105, 15902–15907. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Andres, D.A.; Spielmann, H.P.; Young, S.G.; Fong, L.G. Progerin elicits disease phenotypes of progeria in mice whether or not it is farnesylated. J. Clin Invest. 2008, 118, 3291–3300. [Google Scholar] [CrossRef] [Green Version]
- Gordon, L.B.; Shappell, H.; Massaro, J.; D’Agostino, R.B., Sr.; Brazier, J.; Campbell, S.E.; Kleinman, M.E.; Kieran, M.W. Association of Lonafarnib Treatment vs. No Treatment With Mortality Rate in Patients with Hutchinson–Gilford Progeria Syndrome. JAMA 2018, 319, 1687–1695. [Google Scholar] [CrossRef] [Green Version]
- Gordon, L.B.; Massaro, J.; D’Agostino, R.B., Sr.; Campbell, S.E.; Brazier, J.; Brown, W.T.; Kleinman, M.E.; Kieran, M.W.; Progeria Clinical Trials, C. Impact of farnesylation inhibitors on survival in Hutchinson–Gilford progeria syndrome. Circulation 2014, 130, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, L.B.; Kleinman, M.E.; Miller, D.T.; Neuberg, D.S.; Giobbie-Hurder, A.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.; Snyder, B.D.; et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson–Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, 16666–16671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, L.B.; Kleinman, M.E.; Massaro, J.; D’Agostino, R.B., Sr.; Shappell, H.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.H.; Nazarian, A.; et al. Clinical Trial of the Protein Farnesylation Inhibitors Lonafarnib, Pravastatin, and Zoledronic Acid in Children With Hutchinson–Gilford Progeria Syndrome. Circulation 2016, 134, 114–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takauji, Y.; Wada, T.; Takeda, A.; Kudo, I.; Miki, K.; Fujii, M.; Ayusawa, D. Restriction of protein synthesis abolishes senescence features at cellular and organismal levels. Sci. Rep. 2016, 6, 18722. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5′ to 3′) | Reverse (5′ to 3′) |
|---|---|---|
| HPRT | TAATTGGTGGAGATGATCTCTCAAC | TGCCTGACCAAGGAAAGC |
| IL6 | CACACAGACAGCCACTCACC | CATCCATCTTTTTCAGCCATC |
| IL1b | TACCTGTCCTGCGTGTTGAA | TCTTTGGGTAATTTTTGGGATCT |
| CXCL8 | AGACAGCAGAGCACACAAGC | ATGGTTCCTTCCGGTGGT |
| CCL2 | TCAGCCAGATGCAATCAATG | TCCTGAACCCACTTCTGCTT |
| ICAM1 | TGGTAGCAGCCGCAGTCATA | CTCCTTCCTCTTGGCTTAGT |
| VCAM1 | 5CGCTGACAATGAATCCTGTTAGT | GTATTCTTGGGTGATATGTAGACTTG |
| DDIT3 | AAACGGAAACAGAGTGGTCATTCCCC | GTGGGATTGAGGGTCACATCATTGGCA |
| NOS3 | GTGGCTGTCTGCATGGACCT | CCACGATGGTGACTTTGGCT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bidault, G.; Garcia, M.; Capeau, J.; Morichon, R.; Vigouroux, C.; Béréziat, V. Progerin Expression Induces Inflammation, Oxidative Stress and Senescence in Human Coronary Endothelial Cells. Cells 2020, 9, 1201. https://doi.org/10.3390/cells9051201
Bidault G, Garcia M, Capeau J, Morichon R, Vigouroux C, Béréziat V. Progerin Expression Induces Inflammation, Oxidative Stress and Senescence in Human Coronary Endothelial Cells. Cells. 2020; 9(5):1201. https://doi.org/10.3390/cells9051201
Chicago/Turabian StyleBidault, Guillaume, Marie Garcia, Jacqueline Capeau, Romain Morichon, Corinne Vigouroux, and Véronique Béréziat. 2020. "Progerin Expression Induces Inflammation, Oxidative Stress and Senescence in Human Coronary Endothelial Cells" Cells 9, no. 5: 1201. https://doi.org/10.3390/cells9051201