Next Generation Sequencing of Ancient DNA: Requirements, Strategies and Perspectives


Abstract
:1. Introduction
2. Technical issues
2.1. Sequencing instruments and features
2.2. Selection of instrument
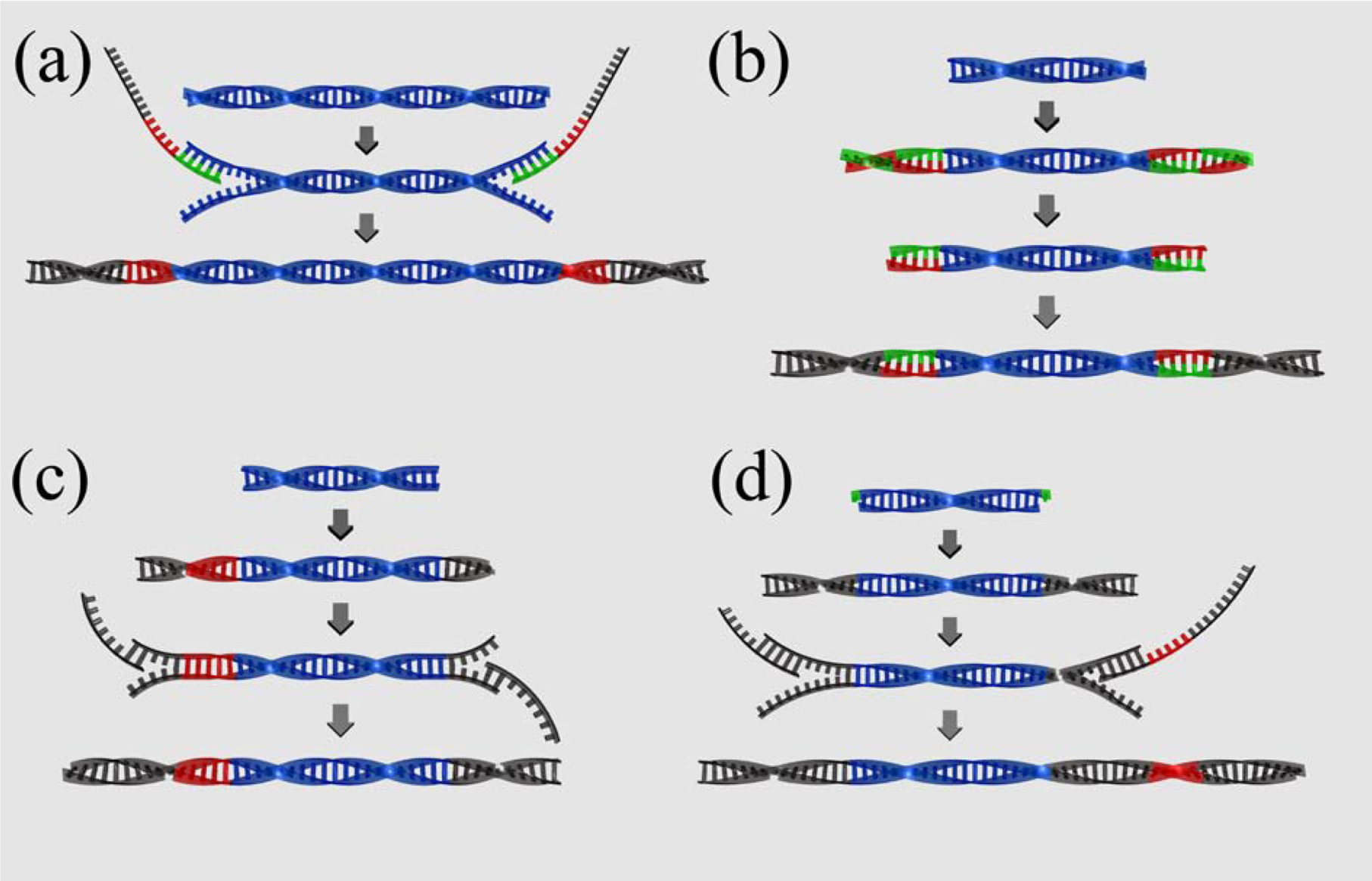
2.3. Library preparation
3. Next generation sequencing of ancient DNA
3.1. Shotgun sequencing
3.2. Target enrichment strategies

{kind=link}
{kind=link}
| Author/Date | Publication Title | Target region | Sequencing/ Target enrichment Strategy |
|---|---|---|---|
| Poinar et al. Jan. 2006 [6] | Metagenomics to paleogenomics: large-scale sequencing of mammoth DNA | genome | shotgun |
| Green et al. Nov. 2006 [24] | Analysis of one million base pairs of Neanderthal DNA | genome | shotgun |
| Gilbert et al. Sep 2007 [25] | Whole-genome shotgun sequencing of mitochondria from ancient hair shafts | mitochondrial genome | shotgun |
| Gilbert et al. Jun. 2008 [26] | Intraspecific phylogenetic analysis of Siberian woolly mammoths using complete mitochondrial genomes | mitochondrial genome | shotgun |
| Gilbert et al. Jun. 2008 [27] | Paleo-Eskimo mtDNA genome reveals matrilineal discontinuity in Greenland | mitochondrial genome | shotgun |
| Green et al. Aug. 2008 [28] | A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing | mitochondrial genome | shotgun |
| Miller et al. Nov. 2008 [8] | Sequencing the nuclear genome of the extinct woolly mammoth | genome | shotgun |
| Miller et al. Feb. 2009 [29] | The mitochondrial genome sequence of the Tasmanian tiger (Thylacinus cynocephalus) | mitochondrial genome | shotgun |
| Allentoft et al. Mar. 2009 [30] | Identification of microsatellites from an extinct moa species using high-throughput (454) sequence data | microsatellites | shotgun |
| Ramírez et al. May 2009 [31] | Paleogenomics in a temperate environment: shotgun sequencing from an extinct Mediterranean caprine | genome | shotgun |
| Willerslev et al. May 2009 [32] | Analysis of complete mitochondrial genomes from extinct and extant rhinoceroses reveals lack of phylogenetic resolution | mitochondrial genome | shotgun |
| Briggs et al. Jul. 2009 [12] | Targeted retrieval and analysis of five Neandertal mtDNA genomes | mitochondrial genome | capture |
| Zhao, Qi and Schuster Aug. 2009 [33] | Tracking the past: interspersed repeats in an extinct Afrotherian mammal, Mammuthus primigenius | genome | shotgun |
| Stiller et al. Oct. 2009 [34] | Direct multiplex sequencing (DMPS)--a novel method for targeted high-throughput sequencing of ancient and highly degraded DNA | mitochondrial genome | multiplex PCR |
| Krause et al. Feb. 2010 [13] | A complete mtDNA genome of an early modern human from Kostenki, Russia | mitochondrial genome | capture |
| Rasmussen et al. Feb. 2010 [10] | Ancient human genome sequence of an extinct Palaeo-Eskimo | genome | shotgun |
| Edwards et al. Feb. 2010 [35] | A complete mitochondrial genome sequence from a mesolithic wild aurochs (Bos primigenius) | mitochondrial genome | shotgun |
| Lindqvist et al. Mar. 2010 [36] | Complete mitochondrial genome of a Pleistocene jawbone unveils the origin of polar bear | mitochondrial genome | shotgun |
| Krause et al. Apr. 2010 [14] | The complete mitochondrial DNA genome of an unknown hominin from southern Siberia | mitochondrial genome | capture |
| Burbano et al. May 2010 [15] | Targeted investigation of the Neandertal genome by array-based sequence capture | nuclear genome | capture |
| Green et al. May 2010 [9] | A draft sequence of the Neandertal genome | genome | shotgun |
1) PCR
2) Hybridization capture

3.3. Barcoding sequencing libraries
1) Roche fusion primers
2) Parallel tagged sequencing

3) Direct multiplex sequencing
4) Illumina barcoding
4. Conclusion
Acknowledgments
References and Notes
- Higuchi, R.; Bowman, B.; Freiberger, M.; Ryder, O.A.; Wilson, A.C. DNA-sequences from the quagga, an extinct member of the horse family. Nature 1984, 312, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Saiki, R.K.; Scharf, S.; Faloona, F.; Mullis, K.B.; Horn, G.T.; Erlich, H.A.; Arnheim, N. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle-cell anemia. Science 1985, 230, 1350–1354. [Google Scholar] [PubMed]
- Mullis, K.B.; Faloona, F.A. Specific synthesis of DNA invitro via a polymerase-catalyzed chain-reaction. Methods Enzymol. 1987, 155, 335–350. [Google Scholar] [PubMed]
- Saiki, R.K.; Gelfand, D.H.; Stoffel, S.; Scharf, S.J.; Higuchi, R.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Primer-directed enzymatic amplification of DNA with a thermostable DNA-polymerase. Science 1988, 239, 487–491. [Google Scholar] [PubMed]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.J.; Chen, Z.T.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [PubMed]
- Poinar, H.N.; Schwarz, C.; Qi, J.; Shapiro, B.; MacPhee, R.D.E.; Buigues, B.; Tikhonov, A.; Huson, D.H.; Tomsho, L.P.; Auch, A.; Rampp, M.; Miller, W.; Schuster, S.C. Metagenomics to paleogenomics: Large-scale sequencing of mammoth DNA. Science 2006, 311, 392–394. [Google Scholar] [CrossRef] [PubMed]
- Noonan, J.P.; Hofreiter, M.; Smith, D.; Priest, J.R.; Rohland, N.; Rabeder, G.; Krause, J.; Detter, J.C.; Pääbo, S.; Rubin, E.M. Genomic sequencing of Pleistocene cave bears. Science 2005, 309, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.; Drautz, D.I.; Ratan, A.; Pusey, B.; Qi, J.; Lesk, A.M.; Tomsho, L.P.; Packard, M.D.; Zhao, F.Q.; Sher, A.; Tikhonov, A.; Raney, B.; Patterson, N.; Lindblad-Toh, K.; Lander, E.S.; Knight, J.R.; Irzyk, G.P.; Fredrikson, K.M.; Harkins, T.T.; Sheridan, S.; Pringle, T.; Schuster, S.C. Sequencing the nuclear genome of the extinct woolly mammoth . Nature 2008, 456, 387–U351. [Google Scholar] [CrossRef] [PubMed]
- Green, R.E.; Krause, J.; Briggs, A.W.; Maricic, T.; Stenzel, U.; Kircher, M.; Patterson, N.; Li, H.; Zhai, W.W.; Fritz, M.H.Y.; et al. A Draft Sequence of the Neandertal Genome. Science 2010, 328, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.; Li, Y.R.; Lindgreen, S.; Pedersen, J.S.; Albrechtsen, A.; Moltke, I.; Metspalu, M.; Metspalu, E.; Kivisild, T.; Gupta, R.; et al. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature 2010, 463, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.B.; Turner, E.H.; Robertson, P.D.; Flygare, S.D.; Bigham, A.W.; Lee, C.; Shaffer, T.; Wong, M.; Bhattacharjee, A.; Eichler, E.E.; Bamshad, M.; Nickerson, D.A.; Shendure, J. Targeted capture and massively parallel sequencing of 12 human exomes . Nature 2009, 461, 272–U153. [Google Scholar] [CrossRef] [PubMed]
- Briggs, A.W.; Good, J.M.; Green, R.E.; Krause, J.; Maricic, T.; Stenzel, U.; Lalueza-Fox, C.; Rudan, P.; Brajković, D.; Kucan, Ž.; Gušić, I.; Schmitz, R.; Doronichev, V.B.; Golovanova, L.V.; de la Rasilla, M.; Fortea, J.; Rosas, A.; Pääbo, S. Targeted Retrieval and Analysis of Five Neandertal mtDNA Genomes . Science 2009, 325, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Krause, J.; Briggs, A.W.; Kircher, M.; Maricic, T.; Zwyns, N.; Derevianko, A.; Pääbo, S. A Complete mtDNA Genome of an Early Modern Human from Kostenki, Russia. Curr. Biol. 2010, 20, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Krause, J.; Fu, Q.M.; Good, J.M.; Viola, B.; Shunkov, M.V.; Derevianko, A.P.; Pääbo, S. The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature 2010, 464, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Burbano, H.A.; Hodges, E.; Green, R.E.; Briggs, A.W.; Krause, J.; Meyer, M.; Good, J.M.; Maricic, T.; Johnson, P.L.F.; Xuan, Z.Y.; Rooks, M.; Bhattacharjee, A.; Brizuela, L.; Albert, F.W.; de la Rasilla, M.; Fortea, J.; Rosas, A.; Lachmann, M.; Hannon, G.J.; Pääbo, S. Targeted Investigation of the Neandertal Genome by Array-Based Sequence Capture. Science 2010, 328, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Millar, C.D.; Huynen, L.; Subramanian, S.; Mohandesan, E.; Lambert, D.M. New developments in ancient genomics. Trends Ecol. Evol. 2008, 23, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Poinar, H.; Kuch, M.; McDonald, G.; Martin, P.; Pääbo, S. Nuclear gene sequences from a late Pleistocene sloth coprolite. Curr. Biol. 2003, 13, 1150–1152. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.M.; Ritchie, P.A.; Millar, C.D.; Holland, B.; Drummond, A.J.; Baroni, C. Rates of evolution in ancient DNA from Adelie penguins. Science 2002, 295, 2270–2273. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, B.; Drummond, A.J.; Rambaut, A.; Wilson, M.C.; Matheus, P.E.; Sher, A.V.; Pybus, O.G.; Gilbert, M.T.P.; Barnes, I.; Binladen, J.; Willerslev, E.; Hansen, A.J.; Baryshnikov, G.F.; Burns, J.A.; Davydov, S.; Driver, J.C.; Froese, D.G.; Harington, C.R.; Keddie, G.; Kosintsev, P.; Kunz, M.L.; Martin, L.D.; Stephenson, R.O.; Storer, J.; Tedford, R.; Zimov, S.; Cooper, A. Rise and fall of the Beringian steppe bison. Science 2004, 306, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Krause, J.; Dear, P.H.; Pollack, J.L.; Slatkin, M.; Spriggs, H.; Barnes, I.; Lister, A.M.; Ebersberger, I.; Pääbo, S.; Hofreiter, M. Multiplex amplification of the mammoth mitochondrial genome and the evolution of Elephantidae. Nature 2006, 439, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Debruyne, R.; Schwarz, C.; Poinar, H. Comment on "Whole-genome shotgun sequencing of mitochondria from ancient hair shafts" . Science 2008, 322, 857b. [Google Scholar] [CrossRef]
- Maricic, T.; Pääbo, S. Optimization of 454 sequencing library preparation from small amounts of DNA permits sequence determination of both DNA strands. Biotechniques 2009, 46, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Kircher, M. Illumina Sequencing Library Preparation for Highly Multiplexed Target Capture and Sequencing . Cold Spring Harb. Protoc. 2010. [Google Scholar] [CrossRef]
- Green, R.E.; Krause, J.; Ptak, S.E.; Briggs, A.W.; Ronan, M.T.; Simons, J.F.; Du, L.; Egholm, M.; Rothberg, J.M.; Paunovic, M.; Pääbo, S. Analysis of one million base pairs of Neanderthal DNA. Nature 2006, 444, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.T.P.; Tomsho, L.P.; Rendulic, S.; Packard, M.; Drautz, D.I.; Sher, A.; Tikhonov, A.; Dalén, L.; Kuznetsova, T.; Kosintsev, P.; Campos, P.F.; Higham, T.; Collins, M.J.; Wilson, A.S.; Shidlovskiy, F.; Buigues, B.; Ericson, P.G.P.; Germonpré, M.; Götherström, A.; Iacumin, P.; Nikolaev, V.; Nowak-Kemp, M.; Willerslev, E.; Knight, J.R.; Irzyk, G.P.; Perbost, C.S.; Fredrikson, K.M.; Harkins, T.T.; Sheridan, S.; Miller, W.; Schuster, S.C. Whole-genome shotgun sequencing of mitochondria from ancient hair shafts. Science 2007, 317, 1927–1930. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.T.P.; Drautz, D.I.; Lesk, A.M.; Ho, S.Y.W.; Qi, J.; Ratan, A.; Hsu, C.H.; Sher, A.; Dalén, L.; Götherström, A.; et al. Intraspecific phylogenetic analysis of Siberian woolly mammoths using complete mitochondrial genomes. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 8327–8332. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.T.P.; Kivisild, T.; Grønnow, B.; Andersen, P.K.; Metspalu, E.; Reidla, M.; Tamm, E.; Axelsson, E.; Götherström, A.; Campos, P.F.; Rasmussen, M.; Metspalu, M.; Higham, T.F.G.; Schwenninger, J.L.; Nathan, R.; De Hoog, C.J.; Koch, A.; Møller, L.N.; Andreasen, C.; Meldgaard, M.; Villems, R.; Bendixen, C.; Willerslev, E. Paleo-Eskimo mtDNA genome reveals matrilineal discontinuity in Greenland. Science 2008, 320, 1787–1789. [Google Scholar] [CrossRef] [PubMed]
- Green, R.E.; Malaspinas, A.S.; Krause, J.; Briggs, A.W.; Johnson, P.L.F.; Uhler, C.; Meyer, M.; Good, J.M.; Maricic, T.; Stenzel, U.; Prüfer, K.; Siebauer, M.; Burbano, H.A.; Ronan, M.; Rothberg, J.M.; Egholm, M.; Rudan, P.; Brajković, D.; Kućan, Ž.; Gušić, I.; Wikström, M.; Laakkonen, L.; Kelso, J.; Slatkin, M.; Pääbo, S. A complete neandertal mitochondrial genome sequence determined by high-throughput Sequencing . Cell 2008, 134, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.; Drautz, D.I.; Janecka, J.E.; Lesk, A.M.; Ratan, A.; Tomsho, L.P.; Packard, M.; Zhang, Y.; McClellan, L.R.; Qi, J.; Zhao, F.; Gilbert, M.T.P.; Dalén, L.; Arsuaga, J.L.; Ericson, P.G.P.; Huson, D.H.; Helgen, K.M.; Murphy, W.J.; Götherström, A.; Schuster, S.C. The mitochondrial genome sequence of the Tasmanian tiger (Thylacinus cynocephalus). Genome Res. 2009, 19, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Allentoft, M.E.; Schuster, S.C.; Holdaway, R.N.; Hale, M.L.; McLay, E.; Oskam, C.; Gilbert, M.T.P.; Spencer, P.; Willerslev, E.; Bunce, M. Identification of microsatellites from an extinct moa species using high-throughput (454) sequence data. Biotechniques 2009, 46, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, O.; Gigli, E.; Bover, P.; Alcover, J.A.; Bertranpetit, J.; Castresana, J.; Lalueza-Fox, C. Paleogenomics in a Temperate Environment: Shotgun Sequencing from an Extinct Mediterranean Caprine . Plos One 2009, 4, e5670. [Google Scholar] [PubMed]
- Willerslev, E.; Gilbert, M.T.P.; Binladen, J.; Ho, S.Y.W.; Campos, P.F.; Ratan, A.; Tomsho, L.P.; da Fonseca, R.R.; Sher, A.; Kuznetsova, T.V.; Nowak-Kemp, M.; Roth, T.L.; Miller, W.; Schuster, S.C. Analysis of complete mitochondrial genomes from extinct and extant rhinoceroses reveals lack of phylogenetic resolution . BMC Evol. Biol. 2009, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.Q.; Qi, J.; Schuster, S.C. Tracking the past: Interspersed repeats in an extinct Afrotherian mammal, Mammuthus primigenius. Genome Res. 2009, 19, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Stiller, M.; Knapp, M.; Stenzel, U.; Hofreiter, M.; Meyer, M. Direct multiplex sequencing (DMPS)-a novel method for targeted high-throughput sequencing of ancient and highly degraded DNA. Genome Res. 2009, 19, 1843–1848. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.J.; Magee, D.A.; Park, S.D.E.; McGettigan, P.A.; Lohan, A.J.; Murphy, A.; Finlay, E.K.; Shapiro, B.; Chamberlain, A.T.; Richards, M.B.; Bradley, D.G.; Loftus, B.J.; MacHugh, D.E. A Complete Mitochondrial Genome Sequence from a Mesolithic Wild Aurochs (Bos primigenius) . Plos One 2010, 5, e9255. [Google Scholar] [PubMed]
- Lindqvist, C.; Schuster, S.C.; Sun, Y.Z.; Talbot, S.L.; Qi, J.; Ratan, A.; Tomsho, L.P.; Kasson, L.; Zeyl, E.; Aars, J.; Miller, W.; Ingólfsson, Ó.; Bachmann, L.; Wiig, Ø. Complete mitochondrial genome of a Pleistocene jawbone unveils the origin of polar bear . Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 5053–5057. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.Y.W.; Gilbert, M.T.P. Ancient mitogenomics. Mitochondrion (Kidlington) 2010, 10, 1–11. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Stenzel, U.; Myles, S.; Prüfer, K.; Hofreiter, M. Targeted high-throughput sequencing of tagged nucleic acid samples . Nucleic Acids Res. 2007, 35, No. 15 e97. [Google Scholar] [CrossRef]
- Römpler, H.; Dear, P.H.; Krause, J.; Meyer, M.; Rohland, N.; Schöneberg, T.; Spriggs, H.; Stiller, M.; Hofreiter, M. Multiplex amplification of ancient DNA. Nat. Protoc. 2006, 1, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Römpler, H.; Rohland, N.; Lalueza-Fox, C.; Willerslev, E.; Kuznetsova, T.; Rabeder, G.; Bertranpetit, J.; Schöneberg, T.; Hofreiter, M. Nuclear gene indicates coat-color polymorphism in mammoths. Science 2006, 313, 62–62. [Google Scholar] [CrossRef] [PubMed]
- Rohland, N.; Malaspinas, A.S.; Pollack, J.L.; Slatkin, M.; Matheus, P.; Hofreiter, M. Proboscidean mitogenomics: Chronology and mode of elephant evolution using mastodon as outgroup. PLoS Biol. 2007, 5, 1663–1671. [Google Scholar] [CrossRef]
- Krause, J.; Unger, T.; Noçon, A.; Malaspinas, A.S.; Kolokotronis, S.O.; Stiller, M.; Soibelzon, L.; Spriggs, H.; Dear, P.H.; Briggs, A.W.; Bray, S.C.E.; O'Brien, S.J.; Rabeder, G.; Matheus, P.; Cooper, A.; Slatkin, M.; Pääbo, S.; Hofreiter, M. Mitochondrial genomes reveal an explosive radiation of extinct and extant bears near the Miocene-Pliocene boundary . BMC Evol. Biol. 2008, 8, 220. [Google Scholar] [CrossRef] [PubMed]
- Prüfer, K.; Stenzel, U.; Hofreiter, M.; Pääbo, S.; Kelso, J.; Green, R.E. Computational challenges in the analysis of ancient DNA . Genome Biology 2010, 11, R47. [Google Scholar] [CrossRef] [PubMed]
- Hofreiter, M.; Jaenicke, V.; Serre, D.; von Haeseler, A.; Pääbo, S. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 2001, 29, 4793–4799. [Google Scholar] [CrossRef] [PubMed]
- Willerslev, E.; Hansen, A.J.; Binladen, J.; Brand, T.B.; Gilbert, M.T.P.; Shapiro, B.; Bunce, M.; Wiuf, C.; Gilichinsky, D.A.; Cooper, A. Diverse plant and animal genetic records from Holocene and Pleistocene sediments. Science 2003, 300, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Hofreiter, M.; Mead, J.I.; Martin, P.; Poinar, H.N. Molecular caving . Curr. Biol. 2003, 13, R693–R695. [Google Scholar] [CrossRef] [PubMed]
- Willerslev, E.; Cappellini, E.; Boomsma, W.; Nielsen, R.; Hebsgaard, M.B.; Brand, T.B.; Hofreiter, M.; Bunce, M.; Poinar, H.N.; Dahl-Jensen, D.; Johnsen, S.; Steffensen, J.P.; Bennike, O.; Schwenninger, J.L.; Nathan, R.; Armitage, S.; de Hoog, C.J.; Alfimov, V.; Christl, M.; Beer, J.; Muscheler, R.; Barker, J.; Sharp, M.; Penkman, K.E.H.; Haile, J.; Taberlet, P.; Gilbert, M.T.P.; Casoli, A.; Campani, E.; Collins, M.J. Ancient biomolecules from deep ice cores reveal a forested Southern Greenland. Science 2007, 317, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Albert, T.J.; Molla, M.N.; Muzny, D.M.; Nazareth, L.; Wheeler, D.; Song, X.Z.; Richmond, T.A.; Middle, C.M.; Rodesch, M.J.; Packard, C.J.; Weinstock, G.M.; Gibbs, R.A. Direct selection of human genomic loci by microarray hybridization. Nat. Meth. 2007, 4, 903–905. [Google Scholar] [CrossRef]
- Hodges, E.; Xuan, Z.; Balija, V.; Kramer, M.; Molla, M.N.; Smith, S.W.; Middle, C.M.; Rodesch, M.J.; Albert, T.J.; Hannon, G.J.; McCombie, W.R. Genome-wide in situ exon capture for selective resequencing. Nat. Genet. 2007, 39, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Gnirke, A.; Melnikov, A.; Maguire, J.; Rogov, P.; LeProust, E.M.; Brockman, W.; Fennell, T.; Giannoukos, G.; Fisher, S.; Russ, C.; Gabriel, S.; Jaffe, D.B.; Lander, E.S.; Nusbaum, C. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009, 27, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Hodges, E.; Rooks, M.; Xuan, Z.Y.; Bhattacharjee, A.; Gordon, D.B.; Brizuela, L.; McCombie, W.R.; Hannon, G.J. Hybrid selection of discrete genomic intervals on custom-designed microarrays for massively parallel sequencing. Nat. Protoc. 2009, 4, 960–974. [Google Scholar] [CrossRef]
- Mamanova, L.; Coffey, A.J.; Scott, C.E.; Kozarewa, I.; Turner, E.H.; Kumar, A.; Howard, E.; Shendure, J.; Turner, D.J. Target-enrichment strategies for next-generation sequencing. Nat. Meth. 2010, 7, 111–118. [Google Scholar] [CrossRef]
- Noonan, J.P.; Coop, G.; Kudaravalli, S.; Smith, D.; Krause, J.; Alessi, J.; Platt, D.; Pääbo, S.; Pritchard, J.K.; Rubin, E.M. Sequencing and analysis of Neanderthal genomic DNA. Science 2006, 314, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Anderung, C.; Persson, P.; Bouwman, A.; Elburg, R.; Götherström, A. Fishing for ancient DNA. Forensic Sci. Int.: Genetics 2008, 2, 104–107. [Google Scholar] [CrossRef]
- Briggs, A.W.; Good, J.M.; Green, R.E.; Krause, J.; Maricic, T.; Stenzel, U.; Pääbo, S. Primer Extension Capture: Targeted Sequence Retrieval from Heavily Degraded DNA Sources . J. Vis. Exp. 2009, 31. [Google Scholar] [CrossRef]
- Meyer, M.; Stenzel, U.; Hofreiter, M. Parallel tagged sequencing on the 454 platform. Nat. Protoc. 2008, 3, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, B.; Hofreiter, M. Analysis of ancient human genomes. Bioessays 2010, 32, 388–391. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Knapp, M.; Hofreiter, M. Next Generation Sequencing of Ancient DNA: Requirements, Strategies and Perspectives. Genes 2010, 1, 227-243. https://doi.org/10.3390/genes1020227
Knapp M, Hofreiter M. Next Generation Sequencing of Ancient DNA: Requirements, Strategies and Perspectives. Genes. 2010; 1(2):227-243. https://doi.org/10.3390/genes1020227
Chicago/Turabian StyleKnapp, Michael, and Michael Hofreiter. 2010. "Next Generation Sequencing of Ancient DNA: Requirements, Strategies and Perspectives" Genes 1, no. 2: 227-243. https://doi.org/10.3390/genes1020227
APA StyleKnapp, M., & Hofreiter, M. (2010). Next Generation Sequencing of Ancient DNA: Requirements, Strategies and Perspectives. Genes, 1(2), 227-243. https://doi.org/10.3390/genes1020227