Sex Differences in the Epigenome: A Cause or Consequence of Sexual Differentiation of the Brain?

1
Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA
2
Watson School of Biological Sciences, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA
*
Author to whom correspondence should be addressed.
Genes 2019, 10(6), 432; https://doi.org/10.3390/genes10060432
Submission received: 20 April 2019
/
Revised: 29 May 2019
/
Accepted: 3 June 2019
/
Published: 7 June 2019
(This article belongs to the Special Issue Epigenetics, Environment, and Brain Disorders)
{kind=link}
{kind=link}
Abstract
:Females and males display differences in neural activity patterns, behavioral responses, and incidence of psychiatric and neurological diseases. Sex differences in the brain appear throughout the animal kingdom and are largely a consequence of the physiological requirements necessary for the distinct roles of the two sexes in reproduction. As with the rest of the body, gonadal steroid hormones act to specify and regulate many of these differences. It is thought that transient hormonal signaling during brain development gives rise to persistent sex differences in gene expression via an epigenetic mechanism, leading to divergent neurodevelopmental trajectories that may underlie sex differences in disease susceptibility. However, few genes with a persistent sex difference in expression have been identified, and only a handful of studies have employed genome-wide approaches to assess sex differences in epigenomic modifications. To date, there are no confirmed examples of gene regulatory elements that direct sex differences in gene expression in the brain. Here, we review foundational studies in this field, describe transcriptional mechanisms that could act downstream of hormone receptors in the brain, and suggest future approaches for identification and validation of sex-typical gene programs. We propose that sexual differentiation of the brain involves self-perpetuating transcriptional states that canalize sex-specific development.
1. Introduction
“In the study of development we are interested not only in the final state to which the system arrives, but also in the course by which it gets there” C.H. Waddington, 1957.
As described in detail [1,2,3], the definition of epigenetics has evolved over time, leading to a relaxed usage of the term, particularly in popular culture (see “From epigenetic landscapes to epigenetic pancakes” at http://blogs.nottingham.ac.uk/makingsciencepublic/). In the study of sex differences—and in the field of neuroepigenetics in general—“epigenetic” refers to covalent DNA or histone modifications that exist “over” DNA and influence gene expression. Frequently, differences in the epigenome, primarily in DNA methylation, are assumed to be causal for phenotypic differences in gene expression, disease incidence, or behavior. However, when Waddington coined the term “epigenetics” in 1942 (republished as Reference [4]), ten years prior to the Hershey-Chase experiments [5], he had no concept of the physical nature of a gene or what we now call the “epigenome”. Rather, he intended to symbolize a developmental process through an “epigenetic landscape” [6], in which each valley represents the segregation of “developmental competence”, or the cell fate decisions made throughout embryogenesis. In Waddington’s landscape, “equilibrium is not centered on a static state, but rather on a direction or pathway of change” [7]. This concept is represented by the term “homeorhesis” (rhesis meaning flow) to distinguish it from the more widely-used “homeostasis”. To date, however, most research has focused on identifying epigenomic endpoints rather than elucidating processes that give rise to such endpoints. Throughout this article, we consider sexual differentiation of the brain as a developmental progression. We suggest that rather than focusing on the epigenomic signature that is a consequence of cell fate decisions, researchers should identify the regulatory mechanisms of transcription factors that establish and/or maintain this signature, as this is crucial for understanding the origin of sex differences in gene expression.
2. Hormone Signaling at Birth Defines Sex Differences in Brain Function
Sex differences in the brain arise from contributions of sex chromosomes and gonadal steroid hormones. The classic Organization and Activation hypothesis, first articulated sixty years ago, states that hormone signaling in early life specifies sex differences in the brain, which are subsequently activated by adult hormones to produce sex-typical reproductive and territorial behaviors [8,9]. At birth, male mice and rats experience a dramatic increase in circulating testosterone that is converted to estradiol directly in the brain by aromatase, a P450 enzyme [10,11]. Estradiol binds its canonical receptors estrogen receptor alpha and beta (ERα/β), which are nuclear receptor transcription factors that are recruited to DNA in response to ligand binding [12]. Estrogens can also act rapidly at the cell membrane to increase neuronal firing [13,14,15,16], potentially initiating activity-dependent transcriptional programs that differ from those directed by nuclear ERα [17]. ERα is thought to be the master regulator of sexual differentiation of the brain. Genetic deletion of this receptor attenuates male-typical sexual and territorial behaviors and feminizes the expression of ERβ and androgen receptor (AR), the receptor for testosterone [18,19,20,21,22]. ERβ also plays critical roles in sexual receptivity, fertility, lactation, and other aspects of female physiology.
Neonatal exposure to estradiol alters the developmental trajectory of the brain, affecting cell number, differentiation, and wiring days after the hormone surge has subsided [23,24]. In males, estradiol induces neurite outgrowth and promotes cell survival in some brain areas, such as the medial amygdala (MeA) and bed nucleus of the stria terminalis (BNST) [25,26], while simultaneously initiating apoptosis in the anteroventral periventricular (AVPV) hypothalamus, a region that regulates ovulation in females [25,27]. Events such as these contribute to sex differences in the function of innate behavioral circuitry. For instance, males and females engage in different mating routines, and only males urine-mark their territory and aggressively defend it, although lactating females will attack intruding conspecifics to defend their offspring [28,29,30,31]. There are also extensive sex differences in stress responses and motivated behaviors [32,33,34]. During estrus, females are sexually receptive to males. Yet if females receive estradiol during the first week of life, adult estrus elicits aggression toward males instead of receptivity [26]. How do females “remember” an estradiol treatment weeks after it has subsided?
The field has long assumed that estradiol directs sexual differentiation of the brain via an epigenetic mechanism, whereby transient exposure to estradiol during a neonatal critical period irreversibly modifies the chromatin state of gene regulatory elements [35,36,37,38,39,40,41], schematized in Figure 1. This process could result in either a constitutive sex difference in gene expression or a sex-specific transcriptional response to a later signaling cue. Much has been written on this subject, for recent reviews see References [42,43,44,45], and transcriptomic analyses in model organisms and humans have identified genes differentially expressed between the brains of the two sexes [17]. However, only a handful of genes have been found to be regulated by neonatal estradiol in the developing male brain, and there are still no examples of sexually dimorphic chromatin states at specific loci that are causal for sex differences in gene expression. In addition, the gene regulatory strategies employed by hormone receptors in the brain remain obscure, as ERα and AR have primarily been studied in human cancer cell lines.
Here we integrate recent findings on epigenetic regulation of gene expression in the brain and nuclear receptor biology to propose a transcriptional model for how neonatal estradiol could specify the development of a male-typical brain. Notably, although Figure 1 depicts a binary choice between two outcomes–female or male–there are multiple sexual differentiation events in the brain. Due to intrinsic transcriptional and epigenetic heterogeneity, different cell types may adopt unique cell fates in response to neonatal estradiol. The aggregation of such cell fates could then produce a spectrum of sexually differentiated phenotypic states. Or, as Waddington said: “The alternative between maleness and femaleness is not so definite… the intersexual condition is no more a case of sharp alternative than is pituitary dwarfism. It becomes merely one of the numerous cases in which the normal well-defined alternatives are disrupted by changes in the genotypic system on which they are based” [6].
3. Regulation of Gene Expression in the Brain
Mammalian gene transcription is directed by transcription factors and influenced by DNA methylation, post-translational modifications (PTMs) to histone proteins, and chromatin organization [46,47,48]. While hormone receptors have been shown to regulate these properties in cell culture models, particularly breast (ERα) and prostate (AR) cancers, few studies have examined how they are regulated in the brain. Moreover, the longevity and renewal of such modifications in post-mitotic neurons remain unclear. In this section, we discuss the mechanisms by which hormone receptors regulate the epigenome and their potential role in the epigenetic maintenance of sex differences in the brain. We review studies on sex differences in the neuronal epigenome in the context of recent findings on the nature of epigenetic regulation of gene expression in the brain. Although there are other factors that contribute to sex differences in the brain, here we focus on ERα, as this receptor is the master regulator of sexual differentiation of the rodent brain, and its role in gene regulation has been extensively studied [17].
3.1. DNA Methylation
The predominant mechanism associated with the epigenetic regulation of gene expression is DNA cytosine methylation (5mC). This modification is relatively stable, correlates with gene repression or activation depending on its genomic location, and is necessary for development, as revealed by mouse genetics [49]. It can maintain genomic imprinting [50], silence transposons [51], and is required for X-inactivation [52]. In the brain, de novo DNA methylation by DNA methyltransferases (DNMTs) is required for learning and memory [53], and there are dynamic changes in DNA methylation in response to neural activity [54,55,56,57]. The mechanism of DNA demethylation in post-mitotic cells was a mystery for years, as no vertebrate homologs to known plant DNA demethylases had been identified [58]. Then in 2009, it was simultaneously published that DNA in the brain contains hydroxymethylated cytosines (5hmC) [59], and that the hydroxylation of 5mC is regulated by the ten-eleven translocation (Tet) family of enzymes, which initiate a cascade that leads to base-excision repair [60]. Further characterization of this pathway led to extensive exploration of stimuli-induced DNA demethylation in the brain [61,62].
Whole genome bisulfite sequencing (WGBS) of neurons has revealed enrichment of DNA methylation on cytosine residues that are not followed by a guanine (denoted as mCH), and that the most common alternate base is an adenine (mCA) [63]. This modification is found in other tissues, but it increases in the brains of mice and humans during postnatal development, concomitant with synaptogenesis and experience-dependent neural activity [63,64,65]. In contrast, mCG patterns are established prenatally and appear stable over time, although it is likely that alterations in mCG within specific circuits or cell types would not be detected in analyses of bulk tissue. WGBS in genetically-defined neuronal types combined with ChIP-seq analysis of Dnmt3a recruitment during postnatal development revealed that mCA is deposited by Dnmt3a over the gene bodies to maintain cell-type specific gene repression [65]. Collectively, this recent work on mCA suggests that this type of DNA methylation plays a crucial role in refining cell identity as neuronal connectivity is established.
Can perinatal estradiol directly alter DNA methylation state and the expression pattern of associated genes? Reports of sex differences in DNA methylation in the brain have assessed either individual methylated cytosines at promoters [66,67,68] or reported percent methylation across the entire genome [69,70]. There remain no examples of differentially-methylated regions that are causally linked to sex differences in gene expression. To date, the best example of the epigenetic effects of perinatal estradiol comes from the gene encoding ERa (Esr1). Esr1 is expressed in the brains of both sexes beginning in mid-gestation [71] and is downregulated by perinatal estradiol via an unknown mechanism [72], leading to increased expression of this receptor in some brain areas in females compared to males [73,74].
There are many reports of sex differences in DNA methylation of the Esr1 promoter [66,67,68,75,76,77,78,79], although the residues identified as differentially hypo- or hyper-methylated vary across studies. In humans, the proximal promoter of human ESR1 is hypomethylated across tissues, suggesting that variation in methylation at individual promoter CpGs is not causal for expression levels of Esr1. In fact, all CpGs at promoters are hypomethylated across the genome, regardless of the level of expression of the associated gene [64]. However, methylation at upstream alternative Esr1 promoters is tissue-specific, consistent with work demonstrating that Esr1 is expressed from these alternate promoters in distinct tissues and tumor types [68,76,80,81]. WGBS across multiple developmental time points could reveal distal regulatory elements that maintain sex differences in the expression of Esr1 and other estrogen-regulated genes [82]. Yet it is still unclear how estrogens would directly alter DNA methylation. There is little evidence of interaction between ERα and DNMT or Tet enzymes, although both DNA methylation and loss of TET2 can attenuate gene activation by ERα in breast cancer cell lines and tumors [83,84,85].
3.2. Histone Modifications
In addition to DNA methylation, chemical modification of amino acid residues primarily within the N-terminal tail of histone proteins has long been associated with epigenetic regulation of gene expression [86]. Such modifications physically alter chromatin structure via electrostatic interactions as well as attract reader proteins that exert diverse functions, such as chromatin remodeling and recruitment of transcriptional machinery [87]. Because certain histone PTMs, either alone or in combination, are strongly associated with distinct classes of cis-regulatory elements (“histone code” hypothesis) and can persist in the absence of an initiating signal, they are often considered an epigenetic modification [86,88,89].
The perinatal hormone surge has been hypothesized to impart such histone PTMs, resulting in chromatin states that maintain sex-specific gene expression programs [38,90]. Although such chromatin states have not been described, there is an extensive characterization of the interaction of ERα with histone acetyltransferases (HATs), methyltransferases, and deacetylases (HDACs), the SWI/SNF nucleosome remodeling complex, and Mediator protein [91]. ERα is tightly associated with steroid receptor coactivators (SRCs), the p160 coactivator, and p300, which has HAT activity and occupies active enhancers [12,92,93]. Broad manipulation of coregulator or HDAC function disrupts brain development [94,95,96,97], but as with DNA methylation, there are few examples of persistent histone modifications or transcription factor occupancy at specific loci [43]. One study evaluated sex differences in trimethylation of lysine 4 on histone H3, a histone modification found at the promoters of active genes, but found little correlation between increased promoter H3K4me3 and gene expression [98].
One common criticism of the role of histone PTMs in epigenetic maintenance is that active chromatin marks are thought to depend on transcriptional activators for renewal [2,99]. In the absence of an epigenetic initiator, histone acetylation has a comparatively short half-life on core histones [100], although H3K4me1 persists longer and is present on poised genes that are transcribed at low levels [101,102,103]. In contrast, histone PTMs associated with repressed chromatin, such as H3K27me3 and H3K9me3, can be generated and recognized by the same protein complex, constituting an intrinsic positive feedback loop. The Polycomb group proteins, which assemble into polycomb repressive complex 1 (PRC1) or 2 (PRC2), fulfill this dual reader-writer function and play well-known roles in stem cell differentiation, as well as cell-type maintenance [104,105]. The involvement of PRC1/2 in maintaining neuron cell fate is an active area of investigation [106,107,108,109]. Although ERα has primarily been shown to recruit co-activator complexes upon ligand binding, it can also recruit co-repressors that are expressed in the brain [91,110]. Thus, neonatal estradiol could impart self-sustaining repressive chromatin in males, resulting in a female-bias in the expression of certain genes or a female-specific transcriptional response to a later signaling cue.
3.3. Genome Organization
Advances in imaging and genomic technologies have made it possible to observe the 3D structure of the genome, revealing that interphase chromosomes are organized into distinct compartments so that active genes are located in the same physical space within the nucleus [111,112]. It is now thought that genome organization itself constitutes an additional level of epigenetic regulation as differentiated cells exhibit unique patterns of chromosomal compartmentalization. Genome organization through interaction with nuclear architecture appears to be of particular importance in neurons, which must maintain a defined cell identity program throughout the life of the organism while retaining the capacity for rapid transcriptional responses to neural activity [113,114,115]. Alterations to nuclear organization can lead to low-level dysregulation of many genes and have been suggested to underlie neurological and psychiatric disorders, as mutations in many nuclear architecture proteins are associated with disease [114,116,117,118,119,120].
Similar to other transcription factors, nuclear hormone receptors regulate gene expression by looping distal enhancer elements and associated regulatory machinery to their target promoters [121,122,123,124,125,126,127]. As hormones can induce the expression of thousands of genes, large-scale rearrangements of chromatin can occur, resulting in significant structural changes in the genome [128,129]. Recent analysis of genome organization in breast cancer cells has revealed large-scale coordinated transcriptional responses to ligand stimulation [130]. Steroid-responsive genes often cluster in groups that are jointly activated or repressed by hormone receptors [122,128,130,131,132]. The acute effects of estradiol on nuclear organization in the brain were first demonstrated in a pioneering electron microscopy study of the ventromedial hypothalamus, which contains many ERα-expressing neurons [133]. The authors implanted gonadectomized female rats with estradiol or sham Silastic capsules and assessed ultrastructural and morphometric nuclear changes. Remarkably, they found that 2 hours of estradiol exposure was sufficient to initiate a robust transcriptional response with significant changes in the size and shape of nuclei, a loss of nuclear envelope invaginations and a decrease in clumped heterochromatin in the nucleoplasm [133]. Taken together, the results from this classic study in the brain combined with more recent work on coordinated gene regulation by ERα in cell lines suggest steroid hormones can “organize” the developing brain by changing the 3D structure of the genome.
4. Sexual Differentiation of the Brain is Developmental Programming
How might sex differences in the epigenome arise throughout development? The simplest and perhaps oldest example of epigenetic regulation is a self-perpetuating transcriptional response that persists in the absence of the original signal [2,134]. When considering sexual differentiation of the brain, it is assumed that sex differences in brain function occur solely from the alteration of the epigenome via covalent modification of DNA or histones. However, cell fate changes are often regulated by the induction of a transcription factor that can then auto-regulate its own expression and establish new cell-type-specific gene expression programs or repress genes associated with alternate fates [135,136,137,138]. In neurodevelopment, this logic has been most clearly elucidated in C. elegans, in which terminal selector transcription factors activate and maintain suites of genes that define differentiated neuron types [139,140].
We propose that developmental hormone signaling directs sexual differentiation of the brain by initiating a positive feedback mechanism that specifies a male-typical cell fate (Figure 2). This process could occur by direct activation of a lineage-defining factor by nuclear hormone receptors, or by the cooperation of these receptors with other transcription factors, as shown in other tissues. Cell-type-specific repertoires of transcription factors and cofactors could then activate or repress distinct cohorts of genes in response to a broad hormone stimulus. Such programs may include genes involved in neurite extension, energy metabolism, synapse formation, or cell adhesion, leading to sex differences in neurodevelopmental trajectories. Importantly, once bound to cis-regulatory elements, lineage-defining factors would recruit cofactors that enzymatically modify chromatin, producing an epigenomic signature that persists in the absence of the original initiating estradiol signal. Although such a signature reflects the activation status of its target genes, it alone does not reveal the epigenetic process responsible for establishing and/or maintaining that gene’s expression pattern.
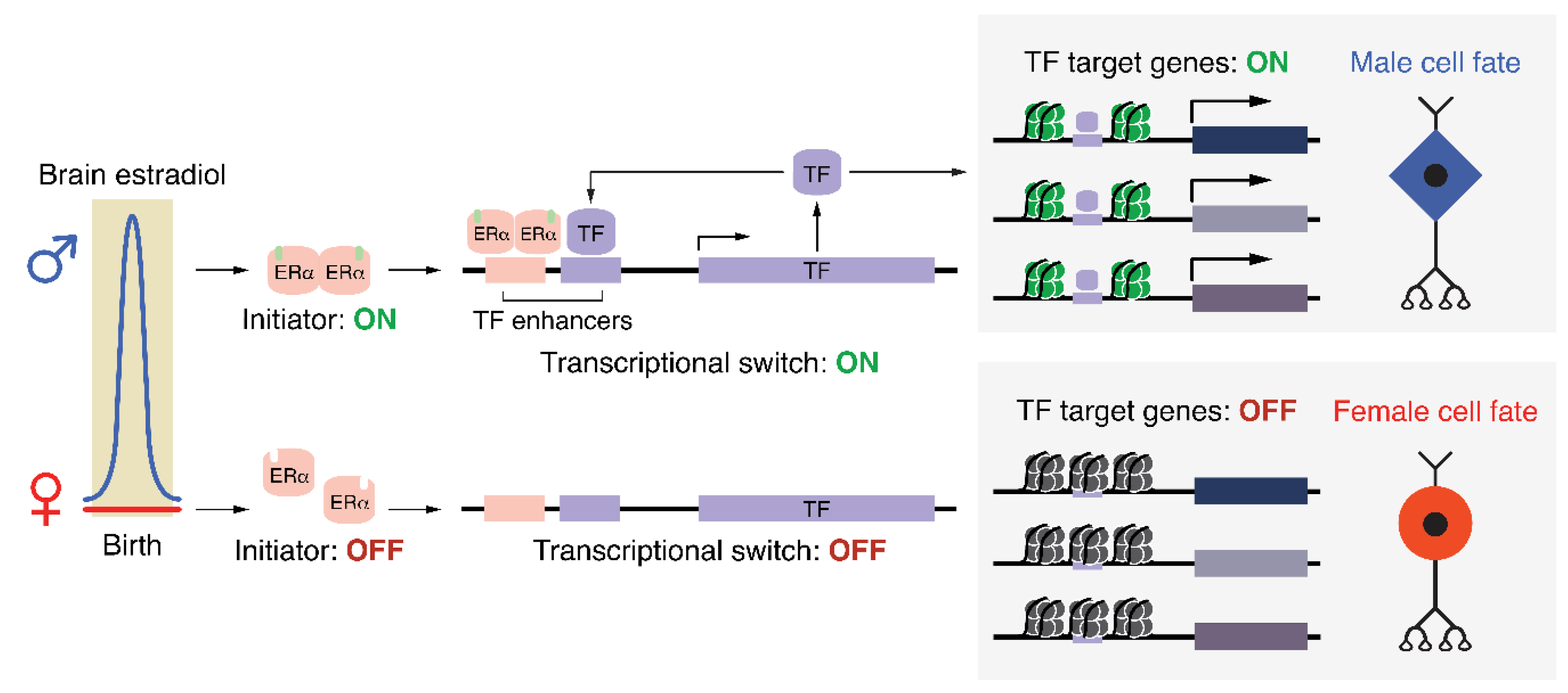
One possible mechanism by which perinatal ERα activation could produce a sex difference in cell fate, as shown thematically in Figure 1. In males (upper), brain estradiol activates a transcriptional initiator (ERα), which turns on the expression of one, or potentially several, transcription factors (TFs). These TFs act as an irreversible ‘transcriptional switch’ by maintaining their expression via positive feedback while also activating genes responsible for producing a male cell fate. Importantly, despite the initiator returning to an inactive state shortly after birth, the ‘transcriptional switch’ remains active, fulfilling Waddington’s definition of an epigenetic process. In females (lower), the lack of estradiol during the perinatal window results in the ‘transcriptional switch’ never being turned on, which, due to the lack of induction of TF target genes, produces the default female cell fate. Epigenomic modifications associated with active or poised genes are schematized as green nucleosomes in males. These same genes in females would reside in a repressed or silenced chromatin state (black nucleosomes).
A conceptual precedent for long-term gene regulation in response to transient hormonal signaling has been observed in D. melanogaster. In this species, the hormone ecdysone, which is released in pulses throughout development, controls the transition from early- to late-born neurons within mushroom bodies (MBs) – structures that process olfactory information. In early-born MB neurons, the transcription factor Chinmo maintains the expression of the ecdysone receptor (EcR-B1). During the larval-pupal transition, a surge of ecdysone activates EcR-B1, which signals for the persistent downregulation of Chinmo and irreversible transition to late-born MB neurons [141]. A similar mechanism occurs in mid-larval neuroblasts, which undergo temporal changes in transcription factor expression to produce different neural cell fates [142]. Indeed, the intersection of steroid hormone release with brain developmental stage varies across species: The critical period for sexual differentiation of the brain can be determined by assessing adult behavioral responses to hormone treatment at different time points. In altricial species that are less developed at birth, the critical period extends into postnatal life, whereas in animals that are more mature at birth, including humans, the critical period occurs during fetal development [143,144].
5. Sex Differences in Gene Regulation May Underlie Sex Differences in Disease Susceptibility
Humans also experience a testosterone surge just after birth, although the role of neonatal hormones in sexual differentiation of the human brain is not known. Rather, a sustained rise in fetal testosterone level during gestational weeks 8–20 is thought to direct masculinization of the brain. Girls with congenital adrenal hyperplasia (CAH) have elevated fetal testosterone and show male-typical levels of childhood aggression, play behaviors, and spatial skills [145,146,147,148]. Throughout the fetal testosterone surge, the brain undergoes extensive neurogenesis and neuronal migration. Although the cell populations expressing hormone receptors and aromatase in the developing human brain have not been identified, it is intriguing to speculate that early testosterone exposure in boys alters the epigenome or chromatin organization of AR target loci, leading to increased variability in gene expression, brain development, and behavior. Identification of hormone-regulated gene programs in the developing brain is likely to reveal sex-specific vulnerabilities: If a neuronal population expresses more of a given gene in females compared to males, females may be less susceptible to heterozygous loss-of-function of that gene. Moreover, genetic variants within non-coding regulatory elements that bind hormone receptors could affect disease susceptibility in a sex-specific manner. A female-specific genetic vulnerability may also result from fluctuating gene expression over the course of the estrous cycle, such a vulnerability could be unmasked by puberty or menopause, both of which are times of increased diagnoses of mental health conditions in women [149,150].
Many psychiatric disorders show a sex bias in incidence and etiology [45,151,152]. A current goal for researchers is to determine whether sex differences in gene expression or regulation can contribute to widely-documented sex differences in disease. Certainly, there are many conditions in which mutations in X-linked genes cause more severe phenotypes in males, which lack an additional functional copy of the gene. However, to the best of our knowledge, there are no examples of sex differences in expression of a particular gene contributing to a corresponding difference in disease incidence or progression. Autism spectrum disorders (ASD) are diagnosed more frequently in boys than girls (4:1), and there is substantial evidence that a genetic component drives this sex-bias [153]. Significant advances in generating functional genomic data from human brains, including ChIP-seq, bisulfite sequencing, and Hi-C, has been transformative for the field of psychiatric genetics. These data allow specific loci to be correlated with gene expression, enhancer activity, and chromatin organization, thereby linking target genes to genetic variants identified in GWAS studies of psychiatric disorders [154,155,156,157,158,159,160,161]. Although no sex differences in regulatory elements have been identified in these studies, they largely focus on cortical areas in which few sex differences in gene expression have been reported in rodents [17]. Interestingly, genes with de novo post-zygotic mutations in individuals with ASD are highly expressed in the fetal amygdala, a region rich in hormone receptors [162]. More extensive epigenomic analysis of human subcortical brain regions that display sex differences in corresponding rodent areas may reveal conserved hormone-regulated gene programs. In the final section of this review, we will discuss experimental approaches and technical innovations that can be applied in mice to identify the transcriptional events that direct sexual differentiation of the brain.
6. Towards a bona fide Epigenetic Mechanism Underlying Sex Differences in the Brain
Do epigenomic sex differences reflect persistent effects of ERs themselves? Or are they a signature of developmental events that occur in one sex but not the other, as a consequence of ER target genes which are no longer active? In order to distinguish between these possibilities, it is necessary to determine the genomic targets of estradiol signaling at birth and to monitor gene expression and chromatin in females, males, and females given estradiol at birth throughout postnatal development. Genes that show increased expression in males and estradiol-treated females at birth may be transient but necessary for masculinization. Similarly, the histone modification H3K27Ac marks active enhancers that will change over time as differentiation and brain wiring progresses. Such experiments would also reveal whether sex differences in gene expression in adulthood are a direct consequence of neonatal or pubertal estradiol signaling or occur via hormone-independent mechanisms.
Studies of gene regulatory mechanisms in the brain have suffered from the same challenges faced by neuroscience in general: How does one achieve spatial and temporal specificity in a highly heterogeneous tissue using mouse genetic tools? Most efforts have employed broad genetic deletion of chromatin modifiers or even pharmacological approaches, which inhibit all histone deacetylation or DNA methylation, to demonstrate the necessity of an individual epigenomic modification. Such brute force approaches will misregulate many genes, not just the one or few of interest, confounding interpretation about the role of an individual locus. One approach to determine a causal role for a regulatory element is to genetically delete that element or target it with a CRISPR/Cas9 system; tethering histone-modifying domains to a catalytically inactive Cas9 is currently the most promising strategy in the brain [163,164,165]. Enhancers are often highly redundant [166], and several may need to be edited at once to achieve an effect; such an experiment has already been performed for ERα enhancers in breast and endometrial cell lines [167].
A common assumption in the study of sex differences is that estrogen-regulated genes contain canonical estrogen receptor response elements (EREs) in their promoters. However, genome-wide studies of ERα binding have revealed that the majority of binding occurs at distal and intronic elements, often via interactions with cell-type-specific TFs [168,169,170]. Accordingly, ERα genomic binding depends on TF co-expression within a particular tissue or cell-type, as shown by comparing RNA-seq and ERα ChIP-seq datasets between mouse liver and aorta, mouse uterus and efferent ductules, and human endometrial and breast cancer cell lines [168,170,171,172]. Such comparisons have implicated broad classes of TF families in context-dependent ERα genomic binding, such as Homeobox (Hox) factors in the uterus [168]. Mass spectrometry-based methods can subsequently identify specific proteins that interact with ERα in different tissues, as done previously in breast cancer cells [173]. To date, however, the genomic binding sites and protein binding partners of ERα have not been examined in brain tissue.
As ERα is recruited to distinct genomic sites in endometrial tissue compared to mammary tissue, there may also be different estrogen-regulated genes in, for example, cortical parvalbumin neurons compared to hippocampal pyramidal neurons or astrocytes. The use of the INTACT (Isolation of Nuclei Tagged in Specific Cell Types) genetic labeling system permits the purification of nuclei from Cre-expressing cells, followed by an assessment of histone modifications or DNA methylation by ChIP-seq or WGBS [65,174]. As sex differences in autosomal gene expression are subtle, the use of INTACT and single-cell methods will facilitate the identification of sex differences in neuronal epigenomes by increasing specificity. Although the enzymes that modify DNA and chromatin are largely ubiquitous, their recruitment to individual regulatory elements often depends on sequence-specific transcription factors. Assessment of transcription factor binding by ChIP-seq in discrete brain regions has been stymied by the high number of cells required for the method. Such experiments are now feasible with the introduction of CUT&RUN, which relies on low cell numbers, potentially even single cells, to identify genomic sites of histone modifications or TF binding [175,176]. Ultimately, it should be possible to determine the genomic binding sites of hormone receptors and other transcription factors in defined neuronal populations. Application of the technical advances described above is likely to reveal the dynamics and mechanisms of the gene regulatory mechanisms that coordinate sexual differentiation of the brain or other critical periods in brain development.
Funding
The authors are funded by the NIH (R01 MH113628 to J.T. and T-32 2T32GM065094 for B.G.), Simons Foundation grant SFARI600568, the Pershing Square Innovation Fund, and the Stanley Family Foundation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Ptashne, M. Epigenetics: Core misconcept. Proc. Natl. Acad. Sci. USA 2013, 110, 7101–7103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henikoff, S.; Greally, J.M. Epigenetics, cellular memory and gene regulation. Curr. Biol. 2016, 26, R644–R648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, S.F. Commentary: “The epigenotype” by C.H. Waddington. Int. J. Epidemiol. 2012, 41, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Hershey, A.D.; Chase, M. Independent functions of viral protein and nucleic acid in growth of bacteriophage. J. Gen. Physiol. 1952, 36, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. Organisers and Genes (1940), by Conrad Hal Waddington; The University Press: Dhaka, Bangladesh, 1940. [Google Scholar]
- Waddington, C.H. The Strategy of the Genes; Routledge: Abingdon-on-Thames, UK, 1957. [Google Scholar]
- Phoenix, C.H.; Goy, R.W.; Gerall, A.A.; Young, W.C. Organizing action of prenatally administered testosterone propionate on the tissues mediating mating behavior in the female guinea pig. Endocrinology 1959, 504, 369–382. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.M.; Wright, C.L.; Schwarz, J.M. New tricks by an old dogma: Mechanisms of the Organizational/Activational Hypothesis of steroid-mediated sexual differentiation of brain and behavior. Horm. Behav. 2009, 55, 655–665. [Google Scholar] [CrossRef]
- Balthazart, J.; Ball, G.F. New insights into the regulation and function of brain estrogen synthase (aromatase). Trends Neurosci. 1998, 21, 243–249. [Google Scholar] [CrossRef]
- Clarkson, J.; Herbison, A.E. Hypothalamic control of the male neonatal testosterone surge. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150115. [Google Scholar] [CrossRef] [Green Version]
- McKenna, N.J.; O’Malley, B.W. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 2002, 108, 465–474. [Google Scholar] [CrossRef]
- Woolley, C.S. Acute Effects of Estrogen on Neuronal Physiology. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 657–680. [Google Scholar] [CrossRef] [PubMed]
- Micevych, P.E.; Kelly, M.J. Membrane estrogen receptor regulation of hypothalamic function. Neuroendocrinology 2012, 96, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Heimovics, S.A.; Trainor, B.C.; Soma, K.K. Rapid Effects of Estradiol on Aggression in Birds and Mice: The Fast and the Furious. Integr. Comp. Biol. 2015, 55, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Stincic, T.L.; Rønnekleiv, O.K.; Kelly, M.J. Diverse actions of estradiol on anorexigenic and orexigenic hypothalamic arcuate neurons. Horm. Behav. 2018, 104, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Gegenhuber, B.; Tollkuhn, J. Signatures of sex: Sex differences in gene expression in the vertebrate brain. Wiley Interdiscip. Rev. Dev. Biol. 2019, e348. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Lubahn, D.B.; Korach, K.S.; Pfaff, D.W. Behavioral effects of estrogen receptor gene disruption in male mice. Proc. Natl. Acad. Sci. USA 1997, 94, 1476–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wersinger, S.R.; Sannen, K.; Villalba, C.; Lubahn, D.B.; Rissman, E.F.; De Vries, G.J. Masculine sexual behavior is disrupted in male and female mice lacking a functional estrogen receptor alpha gene. Horm. Behav. 1997, 32, 176–183. [Google Scholar] [CrossRef]
- Rissman, E.F.; Wersinger, S.R.; Fugger, H.N.; Foster, T.C. Sex with knockout models: Behavioral studies of estrogen receptor. Brain Res. 1999, 835, 80–90. [Google Scholar] [CrossRef]
- Scordalakes, E.M.; Rissman, E.F. Aggression in Male Mice Lacking Functional Estrogen Receptor alpha. Behav. Neurosci. 2003, 117, 38–45. [Google Scholar] [CrossRef]
- Wu, M.V.; Tollkuhn, J. Estrogen receptor alpha is required in GABAergic, but not glutamatergic, neurons to masculinize behavior. Horm. Behav. 2017, 95, 3–12. [Google Scholar] [CrossRef]
- McCarthy, M.M. Estradiol and the developing brain. Physiol. Rev. 2008, 88, 91–124. [Google Scholar] [CrossRef]
- Turano, A.; Osborne, B.F.; Schwarz, J.M. Sexual Differentiation and Sex Differences in Neural Development. Curr. Top. Behav. Neurosci. 2018, 1–42. [Google Scholar] [CrossRef]
- Forger, N.G.; Rosen, G.J.; Waters, E.M.; Jacob, D.; Simerly, R.B.; de Vries, G.J. Deletion of Bax eliminates sex differences in the mouse forebrain. Proc. Natl. Acad. Sci. USA 2004, 101, 13666–13671. [Google Scholar] [CrossRef]
- Wu, M.V.; Manoli, D.S.; Fraser, E.J.; Coats, J.K.; Tollkuhn, J.; Honda, S.-I.; Harada, N.; Shah, N.M. Estrogen masculinizes neural pathways and sex-specific behaviors. Cell 2009, 139, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.A.; Varnum, M.M.; Krentzel, A.A.; Krug, S.; Forger, N.G. Differential control of sex differences in estrogen receptor α in the bed nucleus of the stria terminalis and anteroventral periventricular nucleus. Endocrinology 2013, 154, 3836–3846. [Google Scholar] [CrossRef] [PubMed]
- Beach, F.A. Historical origins of modern research on hormones and behavior. Horm. Behav. 1981, 15, 325–376. [Google Scholar] [CrossRef]
- Lonstein, J.S.; Gammie, S.C. Sensory, hormonal, and neural control of maternal aggression. Neurosci. Biobehav. Rev. 2002, 26, 869–888. [Google Scholar] [CrossRef]
- Hashikawa, K.; Hashikawa, Y.; Lischinsky, J.; Lin, D. The Neural Mechanisms of Sexually Dimorphic Aggressive Behaviors. Trends Genet. 2018, 34, 755–776. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.K.; Touhara, K. Neural circuits regulating sexual behaviors via the olfactory system in mice. Neurosci. Res. 2019, 140, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Bangasser, D.A.; Wicks, B. Sex-specific mechanisms for responding to stress. J. Neurosci. Res. 2017, 95, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Laman-Maharg, A.; Trainor, B.C. Stress, sex, and motivated behaviors. J. Neurosci. Res. 2017, 95, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.B.; Chartoff, E. Sex differences in neural mechanisms mediating reward and addiction. Neuropsychopharmacology 2019, 44, 166–183. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.M.; Auger, A.P.; Bale, T.L.; De Vries, G.J.; Dunn, G.A.; Forger, N.G.; Murray, E.K.; Nugent, B.M.; Schwarz, J.M.; Wilson, M.E. The epigenetics of sex differences in the brain. J. Neurosci. 2009, 29, 12815–12823. [Google Scholar] [CrossRef] [PubMed]
- Bale, T.L.; Baram, T.Z.; Brown, A.S.; Goldstein, J.M.; Insel, T.R.; McCarthy, M.M.; Nemeroff, C.B.; Reyes, T.M.; Simerly, R.B.; Susser, E.S.; et al. Early life programming and neurodevelopmental disorders. Biol. Psychiatry 2010, 68, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Dulac, C. Brain function and chromatin plasticity. Nature 2010, 465, 728–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagnidze, K.; Weil, Z.M.; Pfaff, D.W. Histone modifications proposed to regulate sexual differentiation of brain and behavior. Bioessays 2010, 32, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Auger, A.P.; Jessen, H.M.; Edelmann, M.N. Epigenetic organization of brain sex differences and juvenile social play behavior. Horm. Behav. 2011, 59, 358–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nugent, B.M.; McCarthy, M.M. Epigenetic underpinnings of developmental sex differences in the brain. Neuroendocrinology 2011, 93, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.I.; Mori, H.; Kawata, M. Epigenetic mechanisms are involved in sexual differentiation of the brain. Rev. Endocr. Metab. Disord. 2012, 13, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Hodes, G.E.; Walker, D.M.; Labonté, B.; Nestler, E.J.; Russo, S.J. Understanding the epigenetic basis of sex differences in depression. J. Neurosci. Res. 2017, 95, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Ratnu, V.S.; Emami, M.R.; Bredy, T.W. Genetic and epigenetic factors underlying sex differences in the regulation of gene expression in the brain. J. Neurosci. Res. 2017, 95, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Forger, N.G. Past, present and future of epigenetics in brain sexual differentiation. J. Neuroendocrinol. 2018, 30, e12492. [Google Scholar] [CrossRef] [PubMed]
- Manoli, D.S.; Tollkuhn, J. Gene regulatory mechanisms underlying sex differences in brain development and psychiatric disease. Ann. N. Y. Acad. Sci. 2018, 1420, 26–45. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Shilatifard, A. Chromatin modifications by methylation and ubiquitination: Implications in the regulation of gene expression. Annu. Rev. Biochem. 2006, 75, 243–269. [Google Scholar] [CrossRef] [PubMed]
- Gibcus, J.H.; Dekker, J. The hierarchy of the 3D genome. Mol. Cell 2013, 49, 773–782. [Google Scholar] [CrossRef]
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef] [PubMed]
- Barlow, D.P.; Bartolomei, M.S. Genomic imprinting in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a018382. [Google Scholar] [CrossRef]
- Bestor, T.H.; Bourc’his, D. Transposon silencing and imprint establishment in mammalian germ cells. Cold Spring Harb. Symp. Quant. Biol. 2004, 69, 381–387. [Google Scholar] [CrossRef]
- Chow, J.; Heard, E. X inactivation and the complexities of silencing a sex chromosome. Curr. Opin. Cell Biol. 2009, 21, 359–366. [Google Scholar] [CrossRef]
- Morris, M.J.; Monteggia, L.M. Role of DNA methylation and the DNA methyltransferases in learning and memory. Dialogues Clin. Neurosci. 2014, 16, 359–371. [Google Scholar]
- Chen, W.G. Derepression of BDNF Transcription Involves Calcium-Dependent Phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef]
- Nelson, E.D.; Kavalali, E.T.; Monteggia, L.M. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J. Neurosci. 2008, 28, 395–406. [Google Scholar] [CrossRef]
- Guo, J.U.; Ma, D.K.; Mo, H.; Ball, M.P.; Jang, M.-H.; Bonaguidi, M.A.; Balazer, J.A.; Eaves, H.L.; Xie, B.; Ford, E.; et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 2011, 14, 1345–1351. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Bayraktar, G.; Kreutz, M.R. The Role of Activity-Dependent DNA Demethylation in the Adult Brain and in Neurological Disorders. Front. Mol. Neurosci. 2018, 11, 169. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Mukamel, E. a.; Nery, J.R.; Urich, M.; Puddifoot, C. a.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global Epigenomic Reconfiguration During Mammalian Brain Development. Science 2013, 341, 629–643. [Google Scholar] [CrossRef]
- He, Y.; Ecker, J.R. Non-CG Methylation in the Human Genome. Annu. Rev. Genomics Hum. Genet. 2015, 16, 55–77. [Google Scholar] [CrossRef] [Green Version]
- Stroud, H.; Su, S.C.; Hrvatin, S.; Greben, A.W.; Renthal, W.; Boxer, L.D.; Nagy, M.A.; Hochbaum, D.R.; Kinde, B.; Gabel, H.W.; et al. Early-Life Gene Expression in Neurons Modulates Lasting Epigenetic States. Cell 2017, 171, 1151–1164. [Google Scholar] [CrossRef]
- Kurian, J.R.; Olesen, K.M.; Auger, A.P. Sex differences in epigenetic regulation of the estrogen receptor-alpha promoter within the developing preoptic area. Endocrinology 2010, 151, 2297–2305. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Nugent, B.M.; McCarthy, M.M. Developmental and hormone-induced epigenetic changes to estrogen and progesterone receptor genes in brain are dynamic across the life span. Endocrinology 2010, 151, 4871–4881. [Google Scholar] [CrossRef]
- Westberry, J.M.; Trout, A.L.; Wilson, M.E. Epigenetic regulation of estrogen receptor alpha gene expression in the mouse cortex during early postnatal development. Endocrinology 2010, 151, 731–740. [Google Scholar] [CrossRef]
- Ghahramani, N.M.; Ngun, T.C.; Chen, P.-Y.; Tian, Y.; Krishnan, S.; Muir, S.; Rubbi, L.; Arnold, A.P.; de Vries, G.J.; Forger, N.G.; et al. The effects of perinatal testosterone exposure on the DNA methylome of the mouse brain are late-emerging. Biol. Sex Differ. 2014, 5, 8. [Google Scholar] [CrossRef]
- Nugent, B.M.; Wright, C.L.; Shetty, A.C.; Hodes, G.E.; Lenz, K.M.; Mahurkar, A.; Russo, S.J.; Devine, S.E.; Mccarthy, M.M. Brain feminization requires active repression of masculinization via DNA methylation. Nat. Neurosci. 2015, 18, 690. [Google Scholar] [CrossRef]
- Tobet, S.A.; Henderson, R.G.; Whiting, P.J.; Sieghart, W. Special relationship of gamma-aminobutyric acid to the ventromedial nucleus of the hypothalamus during embryonic development. J. Comp. Neurol. 1999, 405, 88–98. [Google Scholar] [CrossRef]
- DonCarlos, L.L.; McAbee, M.; Ramer-Quinn, D.S.; Stancik, D.M. Estrogen receptor mRNA levels in the preoptic area of neonatal rats are responsive to hormone manipulation. Brain Res. Dev. Brain Res. 1995, 84, 253–260. [Google Scholar] [CrossRef]
- DonCarlos, L.L.; Handa, R.J. Developmental profile of estrogen receptor mRNA in the preoptic area of male and female neonatal rats. Brain Res. Dev. Brain Res. 1994, 79, 283–289. [Google Scholar] [CrossRef]
- Xu, X.; Coats, J.K.; Yang, C.F.; Wang, A.; Ahmed, O.M.; Alvarado, M.; Izumi, T.; Shah, N.M. Modular Genetic Control of Sexually Dimorphic Behaviors. Cell 2012, 148, 596–607. [Google Scholar] [CrossRef] [Green Version]
- Westberry, J.M.; Prewitt, A.K.; Wilson, M.E. Epigenetic regulation of the estrogen receptor alpha promoter in the cerebral cortex following ischemia in male and female rats. Neuroscience 2008, 152, 982–989. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.E.; Westberry, J.M.; Prewitt, A.K. Dynamic regulation of estrogen receptor-alpha gene expression in the brain: A role for promoter methylation? Front. Neuroendocrinol. 2008, 29, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Edelmann, M.N.; Auger, A.P. Epigenetic impact of simulated maternal grooming on estrogen receptor alpha within the developing amygdala. Brain Behav. Immun. 2011, 25, 1299–1304. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.E.; Westberry, J.M.; Trout, A.L. Estrogen receptor-alpha gene expression in the cortex: Sex differences during development and in adulthood. Horm. Behav. 2011, 59, 353–357. [Google Scholar] [CrossRef] [Green Version]
- Westberry, J.M.; Wilson, M.E. Regulation of estrogen receptor alpha gene expression in the mouse prefrontal cortex during early postnatal development. Neurogenetics 2012, 13, 159–167. [Google Scholar] [CrossRef]
- Kos, M.; Reid, G.; Denger, S.; Gannon, F. Minireview: Genomic organization of the human ERalpha gene promoter region. Mol. Endocrinol. 2001, 15, 2057–2063. [Google Scholar] [CrossRef]
- Maekawa, R.; Sato, S.; Okada, M.; Lee, L.; Tamura, I.; Jozaki, K.; Kajimura, T.; Asada, H.; Yamagata, Y.; Tamura, H.; et al. Tissue-Specific Expression of Estrogen Receptor 1 Is Regulated by DNA Methylation in a T-DMR. Mol. Endocrinol. 2016, 30, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Hon, G.C.; Rajagopal, N.; Shen, Y.; McCleary, D.F.; Yue, F.; Dang, M.D.; Ren, B. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat. Genet. 2013, 45, 1198–1206. [Google Scholar] [CrossRef] [Green Version]
- Stone, A.; Zotenko, E.; Locke, W.J.; Korbie, D.; Millar, E.K.A.; Pidsley, R.; Stirzaker, C.; Graham, P.; Trau, M.; Musgrove, E.A.; et al. DNA methylation of oestrogen-regulated enhancers defines endocrine sensitivity in breast cancer. Nat. Commun. 2015, 6, 7758. [Google Scholar] [CrossRef] [Green Version]
- Fleischer, T.; Tekpli, X.; Mathelier, A.; Wang, S.; Nebdal, D.; Dhakal, H.P.; Sahlberg, K.K.; Schlichting, E.; Oslo Breast Cancer Research Consortium (OSBREAC); Børresen-Dale, A.-L.; et al. DNA methylation at enhancers identifies distinct breast cancer lineages. Nat. Commun. 2017, 8, 1379. [Google Scholar] [CrossRef]
- Wang, L.; Ozark, P.A.; Smith, E.R.; Zhao, Z.; Marshall, S.A.; Rendleman, E.J.; Piunti, A.; Ryan, C.; Whelan, A.L.; Helmin, K.A.; et al. TET2 coactivates gene expression through demethylation of enhancers. Sci. Adv. 2018, 4, eaau6986. [Google Scholar] [CrossRef] [Green Version]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Yun, M.; Wu, J.; Workman, J.L.; Li, B. Readers of histone modifications. Cell Res. 2011, 21, 564–578. [Google Scholar] [CrossRef] [Green Version]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Yadav, T.; Quivy, J.-P.; Almouzni, G. Chromatin plasticity: A versatile landscape that underlies cell fate and identity. Science 2018, 361, 1332–1336. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, M.M.; Nugent, B.M. At the frontier of epigenetics of brain sex differences. Front. Behav. Neurosci. 2015, 9, 221. [Google Scholar] [CrossRef]
- Perissi, V.; Rosenfeld, M.G. Controlling nuclear receptors: The circular logic of cofactor cycles. Nat. Rev. Mol. Cell Biol. 2005, 6, 542. [Google Scholar] [CrossRef]
- Foulds, C.E.; Feng, Q.; Ding, C.; Bailey, S.; Hunsaker, T.L.; Malovannaya, A.; Hamilton, R.A.; Gates, L.A.; Zhang, Z.; Li, C.; et al. Proteomic analysis of coregulators bound to ERα on DNA and nucleosomes reveals coregulator dynamics. Mol. Cell 2013, 51, 185–199. [Google Scholar] [CrossRef]
- Yi, P.; Wang, Z.; Feng, Q.; Pintilie, G.D.; Foulds, C.E.; Lanz, R.B.; Ludtke, S.J.; Schmid, M.F.; Chiu, W.; O’Malley, B.W. Structure of a biologically active estrogen receptor-coactivator complex on DNA. Mol. Cell 2015, 57, 1047–1058. [Google Scholar] [CrossRef]
- Apostolakis, E.M.; Ramamurphy, M.; Zhou, D.; Oñate, S.; O’Malley, B.W. Acute disruption of select steroid receptor coactivators prevents reproductive behavior in rats and unmasks genetic adaptation in knockout mice. Mol. Endocrinol. 2002, 16, 1511–1523. [Google Scholar] [CrossRef]
- Molenda, H.A.; Griffin, A.L.; Auger, A.P.; McCarthy, M.M.; Tetel, M.J. Nuclear receptor coactivators modulate hormone-dependent gene expression in brain and female reproductive behavior in rats. Endocrinology 2002, 143, 436–444. [Google Scholar] [CrossRef]
- Murray, E.K.; Hien, A.; de Vries, G.J.; Forger, N.G. Epigenetic control of sexual differentiation of the bed nucleus of the stria terminalis. Endocrinology 2009, 150, 4241–4247. [Google Scholar] [CrossRef]
- Matsuda, K.I.; Mori, H.; Nugent, B.M.; Pfaff, D.W.; McCarthy, M.M.; Kawata, M. Histone deacetylation during brain development is essential for permanent masculinization of sexual behavior. Endocrinology 2011, 152, 2760–2767. [Google Scholar] [CrossRef]
- Shen, E.Y.; Ahern, T.H.; Cheung, I.; Straubhaar, J.; Dincer, A.; Houston, I.; de Vries, G.J.; Akbarian, S.; Forger, N.G. Epigenetics and sex differences in the brain: A genome-wide comparison of histone-3 lysine-4 trimethylation (H3K4me3) in male and female mice. Exp. Neurol. 2015, 268, 21–29. [Google Scholar] [CrossRef]
- Reinberg, D.; Vales, L.D. Chromatin domains rich in inheritance. Science 2018, 361, 33–34. [Google Scholar] [CrossRef]
- Barth, T.K.; Imhof, A. Fast signals and slow marks: The dynamics of histone modifications. Trends Biochem. Sci. 2010, 35, 618–626. [Google Scholar] [CrossRef]
- .Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [Green Version]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann S a Flynn R a Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef]
- Gallegos, D.A.; Chan, U.; Chen, L.-F.; West, A.E. Chromatin Regulation of Neuronal Maturation and Plasticity. Trends Neurosci. 2018, 41, 311–324. [Google Scholar] [CrossRef]
- Boyer, L.A.; Plath, K.; Zeitlinger, J.; Brambrink, T.; Medeiros, L.A.; Lee, T.I.; Levine, S.S.; Wernig, M.; Tajonar, A.; Ray, M.K.; et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006, 441, 349–353. [Google Scholar] [CrossRef]
- Jadhav, U.; Nalapareddy, K.; Saxena, M.; O’Neill, N.K.; Pinello, L.; Yuan, G.-C.; Orkin, S.H.; Shivdasani, R.A. Acquired Tissue-Specific Promoter Bivalency Is a Basis for PRC2 Necessity in Adult Cells. Cell 2016, 165, 1389–1400. [Google Scholar] [CrossRef] [Green Version]
- Lomvardas, S.; Maniatis, T. Histone and DNA Modifications as Regulators of Neuronal Development and Function. Cold Spring Harb. Perspect. Biol. 2016, 8, a024208. [Google Scholar] [CrossRef]
- Feng, X.; Juan, A.H.; Wang, H.A.; Ko, K.D.; Zare, H.; Sartorelli, V. Polycomb Ezh2 controls the fate of GABAergic neurons in the embryonic cerebellum. Development 2016, 143, 1971–1980. [Google Scholar] [CrossRef] [Green Version]
- von Schimmelmann, M.; Feinberg, P.A.; Sullivan, J.M.; Ku, S.M.; Badimon, A.; Duff, M.K.; Wang, Z.; Lachmann, A.; Dewell, S.; Ma’ayan, A.; et al. Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat. Neurosci. 2016, 19, 1321–1330. [Google Scholar] [CrossRef]
- Södersten, E.; Toskas, K.; Rraklli, V.; Tiklova, K.; Björklund, Å.K.; Ringnér, M.; Perlmann, T.; Holmberg, J. A comprehensive map coupling histone modifications with gene regulation in adult dopaminergic and serotonergic neurons. Nat. Commun. 2018, 9, 1226. [Google Scholar] [CrossRef]
- Mahfouz, A.; Lelieveldt, B.P.F.; Grefhorst, A.; van Weert, L.T.C.M.; Mol, I.M.; Sips, H.C.M.; van den Heuvel, J.K.; Datson, N.A.; Visser, J.A.; Reinders, M.J.T.; et al. Genome-wide coexpression of steroid receptors in the mouse brain: Identifying signaling pathways and functionally coordinated regions. Proc. Natl. Acad. Sci. USA 2016, 113, 2738–2743. [Google Scholar] [CrossRef] [Green Version]
- Bickmore, W.A.; Van Steensel, B. Genome architecture: Domain organization of interphase chromosomes. Cell 2013, 152, 1270–1284. [Google Scholar] [CrossRef]
- Dekker, J.; Heard, E. Structural and functional diversity of Topologically Associating Domains. FEBS Lett. 2015, 589, 2877–2884. [Google Scholar] [CrossRef] [Green Version]
- Alexander, J.M.; Lomvardas, S. Nuclear architecture as an epigenetic regulator of neural development and function. Neuroscience 2014, 264, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Medrano-Fernández, A.; Barco, A. Nuclear organization and 3D chromatin architecture in cognition and neuropsychiatric disorders. Mol. Brain 2016, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Watson, L.A.; Tsai, L.H. In the loop: How chromatin topology links genome structure to function in mechanisms underlying learning and memory. Curr. Opin. Neurobiol. 2017, 43, 48–56. [Google Scholar] [CrossRef]
- Rajarajan, P.; Gil, S.E.; Brennand, K.J.; Akbarian, S. Spatial genome organization and cognition. Nat. Rev. Neurosci. 2016, 17, 681–691. [Google Scholar] [CrossRef] [Green Version]
- Rajarajan, P.; Borrman, T.; Liao, W.; Schrode, N.; Flaherty, E.; Casiño, C.; Powell, S.; Yashaswini, C.; LaMarca, E.A.; Kassim, B.; et al. Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science 2018, 362, eaat4311. [Google Scholar] [CrossRef]
- Sun, J.H.; Zhou, L.; Emerson, D.J.; Phyo, S.A.; Titus, K.R.; Gong, W.; Gilgenast, T.G.; Beagan, J.A.; Davidson, B.L.; Tassone, F.; et al. Disease-Associated Short Tandem Repeats Co-localize with Chromatin Domain Boundaries. Cell 2018, 175, 224–238.e15. [Google Scholar] [CrossRef] [Green Version]
- Demontis, D.; Walters, R.K.; Martin, J.; Mattheisen, M.; Als, T.D.; Agerbo, E.; Baldursson, G.; Belliveau, R.; Bybjerg-Grauholm, J.; Bækvad-Hansen, M.; et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet. 2019, 51, 63–75. [Google Scholar] [CrossRef]
- Fudenberg, G.; Pollard, K.S. Chromatin features constrain structural variation across evolutionary timescales. Proc. Natl. Acad. Sci. USA 2019, 116, 2175–2180. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Carroll, J.S.; Brown, M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol. Cell 2005, 19, 631–642. [Google Scholar] [CrossRef]
- Hsu, P.Y.; Hsu, H.K.; Singer, G.A.C.; Yan, P.S.; Rodriguez, B.A.T.; Liu, J.C.; Weng, Y.I.; Deatherage, D.E.; Chen, Z.; Pereira, J.S.; et al. Estrogen-mediated epigenetic repression of large chromosomal regions through DNA looping. Genome Res. 2010, 20, 733–744. [Google Scholar] [CrossRef] [Green Version]
- Fullwood, M.J.; Liu, M.H.; Pan, Y.F.; Liu, J.; Xu, H.; Mohamed, Y.B.; Orlov, Y.L.; Velkov, S.; Ho, A.; Mei, P.H.; et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 2009, 462, 58–64. [Google Scholar] [CrossRef]
- Ross-Innes, C.S.; Brown, G.D.; Carroll, J.S. A co-ordinated interaction between CTCF and ER in breast cancer cells. BMC Genomics 2011, 12, 593. [Google Scholar] [CrossRef]
- Hsieh, C.-L.; Fei, T.; Chen, Y.; Li, T.; Gao, Y.; Wang, X.; Sun, T.; Sweeney, C.J.; Lee, G.-S.M.; Chen, S.; et al. Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc. Natl. Acad. Sci. USA 2014, 111, 7319–7324. [Google Scholar] [CrossRef] [Green Version]
- Panigrahi, A.K.; Foulds, C.E.; Lanz, R.B.; Hamilton, R.A.; Yi, P.; Lonard, D.M.; Tsai, M.-J.; Tsai, S.Y.; O’Malley, B.W. SRC-3 Coactivator Governs Dynamic Estrogen-Induced Chromatin Looping Interactions during Transcription. Mol. Cell 2018, 70, 679–694.e7. [Google Scholar] [CrossRef] [Green Version]
- Le Dily, F.; Vidal, E.; Cuartero, Y.; Quilez, J.; Nacht, A.S.; Vicent, G.P.; Carbonell-Caballero, J.; Sharma, P.; Villanueva-Cañas, J.L.; Ferrari, R.; et al. Hormone-control regions mediate steroid receptor-dependent genome organization. Genome Res. 2019, 29, 29–39. [Google Scholar] [CrossRef]
- Rafique, S.; Thomas, J.S.; Sproul, D.; Bickmore, W.A. Estrogen-induced chromatin decondensation and nuclear re-organization linked to regional epigenetic regulation in breast cancer. Genome Biol. 2015, 16, 145. [Google Scholar] [CrossRef] [Green Version]
- Jubb, A.W.; Boyle, S.; Hume, D.A.; Bickmore, W.A. Glucocorticoid Receptor Binding Induces Rapid and Prolonged Large-Scale Chromatin Decompaction at Multiple Target Loci. Cell Rep. 2017, 21, 3022–3031. [Google Scholar] [CrossRef] [Green Version]
- Le Dily, F.; Beato, M. Signaling by steroid hormones in the 3D nuclear space. Int. J. Mol. Sci. 2018, 19, 306. [Google Scholar] [CrossRef]
- Wiench, M.; Miranda, T.B.; Hager, G.L. Control of nuclear receptor function by local chromatin structure. FEBS J. 2011, 278, 2211–2230. [Google Scholar] [CrossRef] [Green Version]
- Quintin, J.; Le Péron, C.; Palierne, G.; Bizot, M.; Cunha, S.; Sérandour, A.A.; Avner, S.; Henry, C.; Percevault, F.; Belaud-Rotureau, M.-A.; et al. Dynamic estrogen receptor interactomes control estrogen-responsive trefoil Factor (TFF) locus cell-specific activities. Mol. Cell Biol. 2014, 34, 2418–2436. [Google Scholar] [CrossRef]
- Jones, K.J.; Pfaff, D.W.; McEwen, B.S. Early estrogen-induced nuclear changes in rat hypothalamic ventromedial neurons: An ultrastructural and morphometric analysis. J. Comp. Neurol. 1985, 239, 255–266. [Google Scholar] [CrossRef]
- Bonasio, R.; Tu, S.; Reinberg, D. Molecular signals of epigenetic states. Science 2010, 330, 612–616. [Google Scholar] [CrossRef]
- Weintraub, H.; Davis, R.; Krause, S.M.; Benezra, R.; Rupp, R.; Hollenberg, S.; Zhuang, Y.; Lassar, A. The myoD Gene Family: Nodal Point During Specification of the Muscle Cell Lineage. Nature 1987, 335, 155. [Google Scholar] [CrossRef]
- Blau, H.M.; Baltimore, D. Differentiation requires continuous regulation. J. Cell Biol. 1991, 112, 781–783. [Google Scholar] [CrossRef]
- Chen, Z.F.; Paquette, A.J.; Anderson, D.J. NRSF/REST is required in vivo for repression of multiple neuronal target genes during embryogenesis. Nat. Genet. 1998, 20, 136–142. [Google Scholar] [CrossRef]
- Sánchez Alvarado, A.; Yamanaka, S. Rethinking differentiation: Stem cells, regeneration, and plasticity. Cell 2014, 157, 110–119. [Google Scholar] [CrossRef]
- Hobert, O. Regulation of terminal differentiation programs in the nervous system. Annu. Rev. Cell Dev. Biol. 2011, 27, 681–696. [Google Scholar] [CrossRef]
- Hobert, O. Terminal Selectors of Neuronal Identity. Curr. Top. Dev. Biol. 2016, 116, 455–475. [Google Scholar]
- Marchetti, G.; Tavosanis, G. Steroid Hormone Ecdysone Signaling Specifies Mushroom Body Neuron Sequential Fate via Chinmo. Curr. Biol. 2017, 27, 3017–3024. [Google Scholar] [CrossRef]
- Syed, M.H.; Mark, B.; Doe, C.Q. Steroid hormone induction of temporal gene expression in Drosophila brain neuroblasts generates neuronal and glial diversity. Elife 2017, 6, e26287. [Google Scholar] [CrossRef]
- MacLusky, N.J.; Naftolin, F. Sexual Differentiation of the Central Nervous System. Science 1981, 211, 1294–1303. [Google Scholar] [CrossRef]
- Workman, A.D.; Charvet, C.J.; Clancy, B.; Darlington, R.B.; Finlay, B.L. Modeling transformations of neurodevelopmental sequences across mammalian species. J. Neurosci. 2013, 33, 7368–7383. [Google Scholar] [CrossRef]
- Hines, M. Prenatal testosterone and gender-related behaviour. Eur. J. Endocrinol. 2006, 155, S115–S121. [Google Scholar] [CrossRef] [Green Version]
- Hines, M. Sex-related variation in human behavior and the brain. Trends Cogn. Sci. 2010, 14, 448–456. [Google Scholar] [CrossRef] [Green Version]
- Berenbaum, S.A.; Bryk, K.L.K.; Beltz, A.M. Early androgen effects on spatial and mechanical abilities: Evidence from congenital adrenal hyperplasia. Behav. Neurosci. 2012, 126, 86–96. [Google Scholar] [CrossRef]
- Hines, M.; Pasterski, V.; Spencer, D.; Neufeld, S.; Patalay, P.; Hindmarsh, P.C.; Hughes, I.A.; Acerini, C.L. Prenatal androgen exposure alters girls’ responses to information indicating gender-appropriate behaviour. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150125. [Google Scholar] [CrossRef]
- Häfner, H. Gender differences in schizophrenia. Psychoneuroendocrinology 2003, 28, 17–54. [Google Scholar] [CrossRef]
- Schulz, K.M.; Sisk, C.L. The organizing actions of adolescent gonadal steroid hormones on brain and behavioral development. Neurosci. Biobehav. Rev. 2016, 70, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.B.; Berkley, K.J.; Geary, N.; Hampson, E.; Herman, J.P.; Young, E. Sex Differences in the Brain: From Genes to Behavior; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- McCarthy, M.M.; Arnold, A.P.; Ball, G.F.; Blaustein, J.D.; De Vries, G.J. Sex differences in the brain: The not so inconvenient truth. J. Neurosci. 2012, 32, 2241–2247. [Google Scholar] [CrossRef]
- Werling, D.M. The role of sex-differential biology in risk for autism spectrum disorder. Biol. Sex Differ. 2016, 7, 58. [Google Scholar] [CrossRef]
- Doan, R.N.; Bae, B.-I.; Cubelos, B.; Chang, C.; Hossain, A.A.; Al-Saad, S.; Mukaddes, N.M.; Oner, O.; Al-Saffar, M.; Balkhy, S.; et al. Mutations in Human Accelerated Regions Disrupt Cognition and Social Behavior. Cell 2016, 167, 341–354.e12. [Google Scholar] [CrossRef] [Green Version]
- Parikshak, N.N.; Swarup, V.; Belgard, T.G.; Irimia, M.; Ramaswami, G.; Gandal, M.J.; Hartl, C.; Leppa, V.; Ubieta, L.; de la, T.; Huang, J.; et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 2016, 540, 423–427. [Google Scholar] [CrossRef]
- Girdhar, K.; Hoffman, G.E.; Jiang, Y.; Brown, L.; Kundakovic, M.; Hauberg, M.E.; Francoeur, N.J.; Wang, Y.-C.; Shah, H.; Kavanagh, D.H.; et al. Cell-specific histone modification maps in the human frontal lobe link schizophrenia risk to the neuronal epigenome. Nat. Neurosci. 2018, 21, 1126–1136. [Google Scholar] [CrossRef]
- Li, M.; Santpere, G.; Imamura Kawasawa, Y.; Evgrafov, O.V.; Gulden, F.O.; Pochareddy, S.; Sunkin, S.M.; Li, Z.; Shin, Y.; Zhu, Y.; et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 2018, 362, eaat7615. [Google Scholar] [CrossRef]
- de la Torre-Ubieta, L.; Stein, J.L.; Won, H.; Opland, C.K.; Liang, D.; Lu, D.; Geschwind, D.H. The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell 2018, 172, 289–304.e18. [Google Scholar] [CrossRef] [Green Version]
- Short, P.J.; McRae, J.F.; Gallone, G.; Sifrim, A.; Won, H.; Geschwind, D.H.; Wright, C.F.; Firth, H.V.; FitzPatrick, D.R.; Barrett, J.C.; et al. De novo mutations in regulatory elements in neurodevelopmental disorders. Nature 2018, 555, 611–616. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, P.F.; Agrawal, A.; Bulik, C.M.; Andreassen, O.A.; Børglum, A.D.; Breen, G.; Cichon, S.; Edenberg, H.J.; Faraone, S.V.; Gelernter, J.; et al. Psychiatric Genomics: An Update and an Agenda. Am. J. Psychiatry 2018, 175, 15–27. [Google Scholar] [CrossRef]
- Wang, D.; Liu, S.; Warrell, J.; Won, H.; Shi, X.; Navarro, F.C.P.; Clarke, D.; Gu, M.; Emani, P.; Yang, Y.T.; et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 2018, 362, eaat8464. [Google Scholar] [CrossRef]
- Lim, E.T.; Uddin, M.; De Rubeis, S.; Chan, Y.; Kamumbu, A.S.; Zhang, X.; D, A.M.; Kim, S.N.; Sean Hill, R.; Goldberg, A.P.; et al. Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat. Neurosci. 2017, 20, 1217. [Google Scholar] [CrossRef]
- Yang, M.G.; West, A.E. Editing the Neuronal Genome: A CRISPR View of Chromatin Regulation in Neuronal Development, Function, and Plasticity. Yale J. Biol. Med. 2016, 89, 457–470. [Google Scholar]
- Savell, K.E.; Day, J.J. Applications of CRISPR/Cas9 in the Mammalian Central Nervous System. Yale J. Biol. Med. 2017, 90, 567–581. [Google Scholar]
- Savell, K.E.; Bach, S.V.; Zipperly, M.E.; Revanna, J.S.; Goska, N.A.; Tuscher, J.J.; Duke, C.G.; Sultan, F.A.; Burke, J.N.; Williams, D.; et al. A Neuron-Optimized CRISPR/dCas9 Activation System for Robust and Specific Gene Regulation. eNeuro 2019, 6. [Google Scholar] [CrossRef]
- Sabarís, G.; Laiker, I.; Preger-Ben Noon, E.; Frankel, N. Actors with Multiple Roles: Pleiotropic Enhancers and the Paradigm of Enhancer Modularity. Trends Genet. 2019, 35, 423–433. [Google Scholar] [CrossRef]
- Carleton, J.B.; Berrett, K.C.; Gertz, J. Multiplex Enhancer Interference Reveals Collaborative Control of Gene Regulation by Estrogen Receptor α-Bound Enhancers. Cell Syst. 2017, 5, 333–344.e5. [Google Scholar] [CrossRef]
- Hewitt, S.C.; Li, L.; Grimm, S.A.; Chen, Y.; Liu, L.; Li, Y.; Bushel, P.R.; Fargo, D.; Korach, K.S. Research resource: Whole-genome estrogen receptor α binding in mouse uterine tissue revealed by ChIP-seq. Mol. Endocrinol. 2012, 26, 887–898. [Google Scholar] [CrossRef]
- Jozwik, K.M.; Carroll, J.S. Pioneer factors in hormone-dependent cancers. Nat. Rev. Cancer 2012, 12, 381–385. [Google Scholar] [CrossRef]
- Gertz, J.; Savic, D.; Varley, K.E.; Partridge, E.C.; Safi, A.; Jain, P.; Cooper, G.M.; Reddy, T.E.; Crawford, G.E.; Myers, R.M. Distinct properties of cell-type-specific and shared transcription factor binding sites. Mol. Cell. 2013, 52, 25–36. [Google Scholar] [CrossRef]
- Gordon, F.K.; Vallaster, C.S.; Westerling, T.; Iyer, L.K.; Brown, M.; Schnitzler, G.R. Research resource: Aorta- and liver-specific ERα-binding patterns and gene regulation by estrogen. Mol. Endocrinol. 2014, 28, 1337–1351. [Google Scholar] [CrossRef]
- Yao, G.; Hu, S.; Yu, L.; Ru, Y.; Chen, C.D.; Liu, Q.; Zhang, Y. Genome-Wide Mapping of In Vivo ERα-Binding Sites in Male Mouse Efferent Ductules. Endocrinology 2017, 158, 3724–3737. [Google Scholar] [CrossRef]
- Mohammed, H.; D’Santos, C.; Serandour, A.A.; Ali, H.R.; Brown, G.D.; Atkins, A.; Rueda, O.M.; Holmes, K.A.; Theodorou, V.; Robinson, J.L.L.; et al. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep. 2013, 3, 342–349. [Google Scholar] [CrossRef]
- Mo, A.; Mukamel, E.A.; Davis, F.P.; Luo, C.; Henry, G.L.; Picard, S.; Urich, M.A.; Nery, J.R.; Sejnowski, T.J.; Lister, R.; et al. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 2015, 86, 1369–1384. [Google Scholar] [CrossRef] [Green Version]
- Skene, P.J.; Henikoff, J.G.; Henikoff, S. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc. 2018, 13, 1006–1019. [Google Scholar] [CrossRef]
- Hainer, S.J.; Bošković, A.; McCannell, K.N.; Rando, O.J.; Fazzio, T.G. Profiling of Pluripotency Factors in Single Cells and Early Embryos. Cell 2019, 177, 1319–1329. [Google Scholar] [CrossRef]
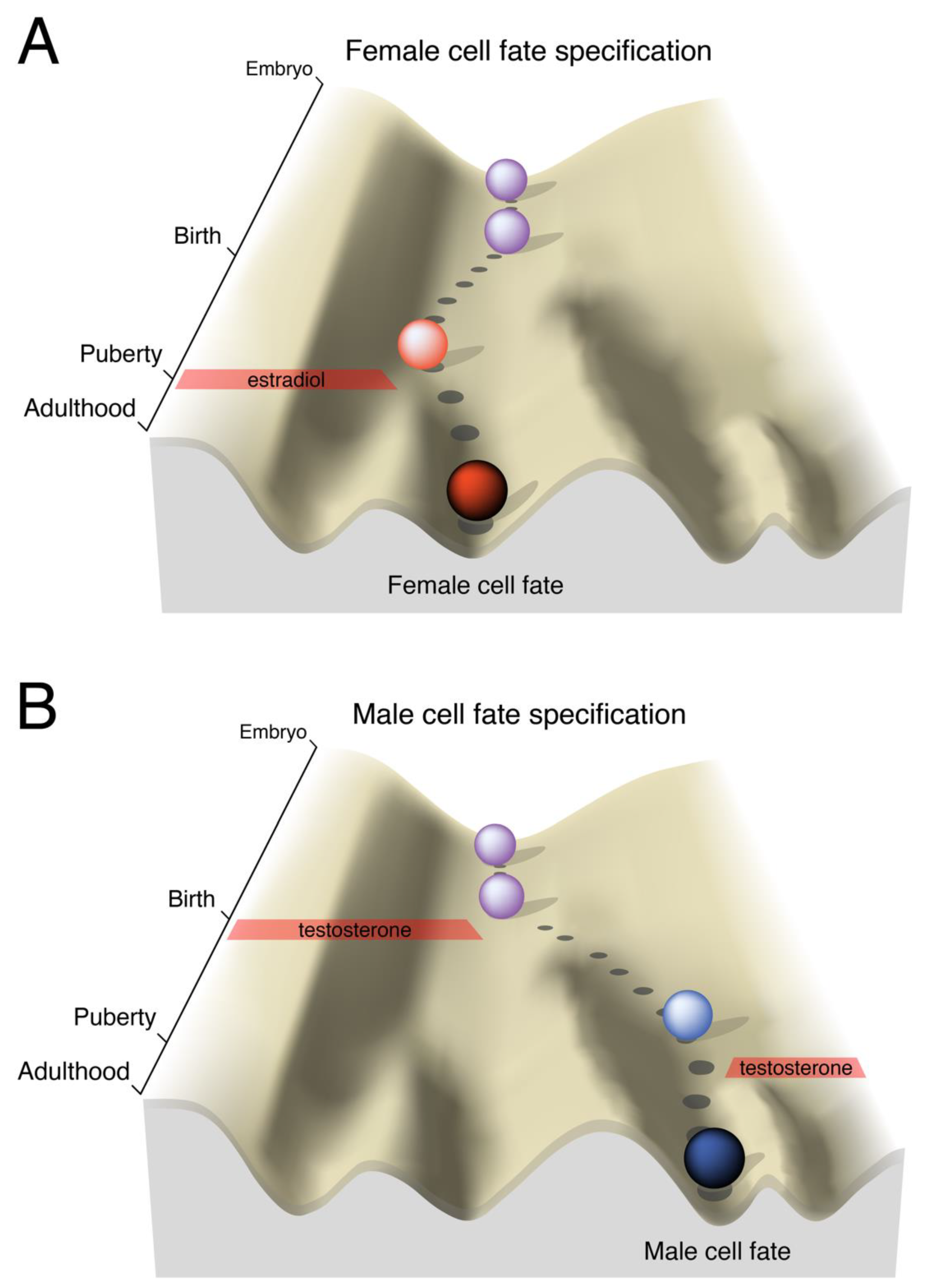
Figure 1.
Developmental landscapes of brain cell fate specification. In Waddington’s description of the epigenetic landscape [6,7], the ball rolling down the hill represents the developmental trajectory of an embryo: The landscape itself is pre-specified by the genetic makeup of the organism, but the chosen path can be altered by extrinsic events. As development progresses, the valley slopes steepen, representing the inevitable canalization of a genetic program as cells differentiate and their plasticity is restricted. Near birth, the rodent brain is bipotential and can easily progress to a female or male fate. A) The brain, like the gonads, develops by default as female. B) In males, testosterone is converted to estradiol in the brain and activates a gene expression program that irreversibly pushes the brain towards a male fate. In both sexes, puberty is an additional sensitive period when sexually differentiated circuitry is acted on by sex-specific hormonal profiles.
Figure 1.
Developmental landscapes of brain cell fate specification. In Waddington’s description of the epigenetic landscape [6,7], the ball rolling down the hill represents the developmental trajectory of an embryo: The landscape itself is pre-specified by the genetic makeup of the organism, but the chosen path can be altered by extrinsic events. As development progresses, the valley slopes steepen, representing the inevitable canalization of a genetic program as cells differentiate and their plasticity is restricted. Near birth, the rodent brain is bipotential and can easily progress to a female or male fate. A) The brain, like the gonads, develops by default as female. B) In males, testosterone is converted to estradiol in the brain and activates a gene expression program that irreversibly pushes the brain towards a male fate. In both sexes, puberty is an additional sensitive period when sexually differentiated circuitry is acted on by sex-specific hormonal profiles.

Figure 2.
Proposed positive feedback mechanism responsible for maintaining sex-specific cell fates. ERα: estrogen receptor alpha; TF: transcription factor.
Figure 2.
Proposed positive feedback mechanism responsible for maintaining sex-specific cell fates. ERα: estrogen receptor alpha; TF: transcription factor.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gegenhuber, B.; Tollkuhn, J. Sex Differences in the Epigenome: A Cause or Consequence of Sexual Differentiation of the Brain? Genes 2019, 10, 432. https://doi.org/10.3390/genes10060432
AMA Style
Gegenhuber B, Tollkuhn J. Sex Differences in the Epigenome: A Cause or Consequence of Sexual Differentiation of the Brain? Genes. 2019; 10(6):432. https://doi.org/10.3390/genes10060432
Chicago/Turabian StyleGegenhuber, Bruno, and Jessica Tollkuhn. 2019. "Sex Differences in the Epigenome: A Cause or Consequence of Sexual Differentiation of the Brain?" Genes 10, no. 6: 432. https://doi.org/10.3390/genes10060432
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.