The Gene Master Regulators (GMR) Approach Provides Legitimate Targets for Personalized, Time-Sensitive Cancer Gene Therapy



Abstract
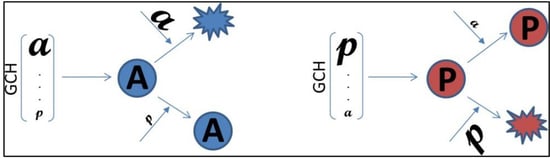
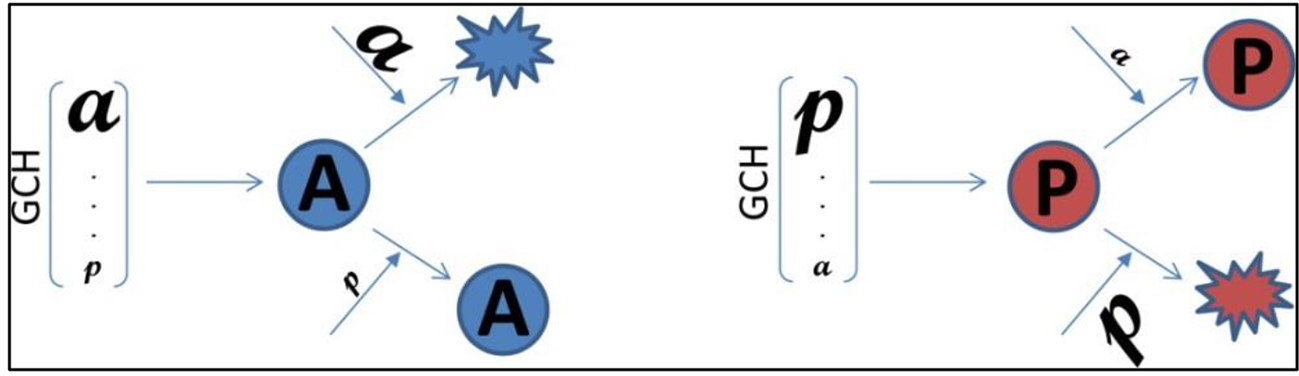
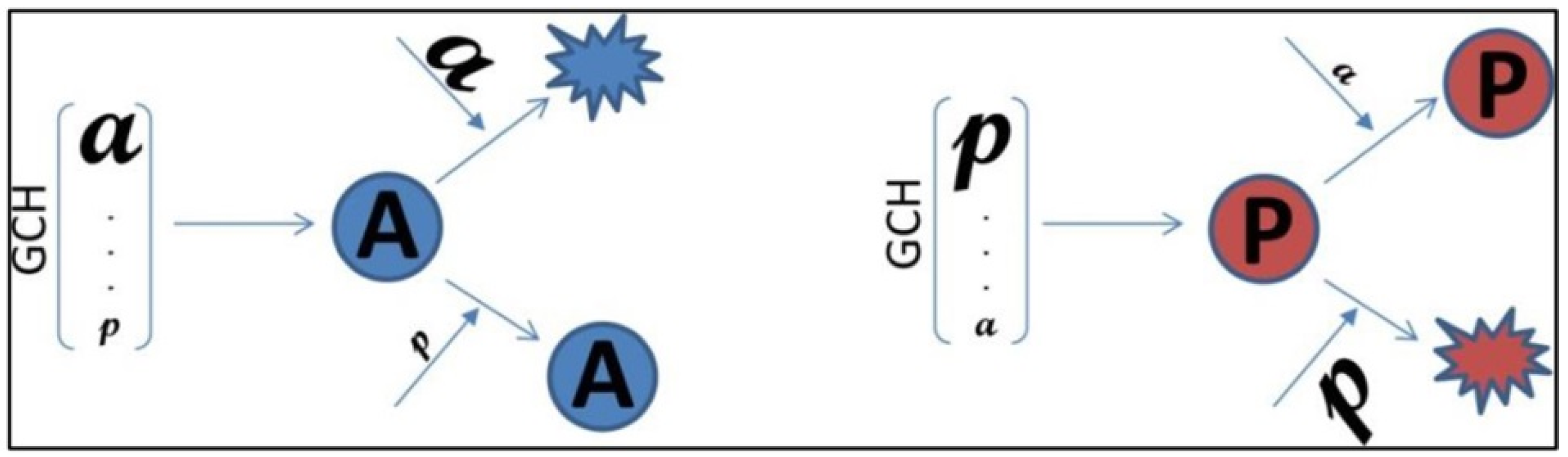
1. Introduction
2. Materials and Methods
2.1. Tumor Samples
2.2. Cell Lines
2.3. Biological Replicas
2.4. Microarray
2.5. Relative Expression Variation (REV) and Relative Expression Stability (RES)
2.6. Expression Regulation
2.7. Expression Correlation
2.8. Gene Commanding Height (GCH)
2.9. Gene Ontology and Functional Pathways
2.10. CANCER-GMR Software
2.11. Experimental Design to Validate the GMR Theory
3. Results
3.1. Experimental Data
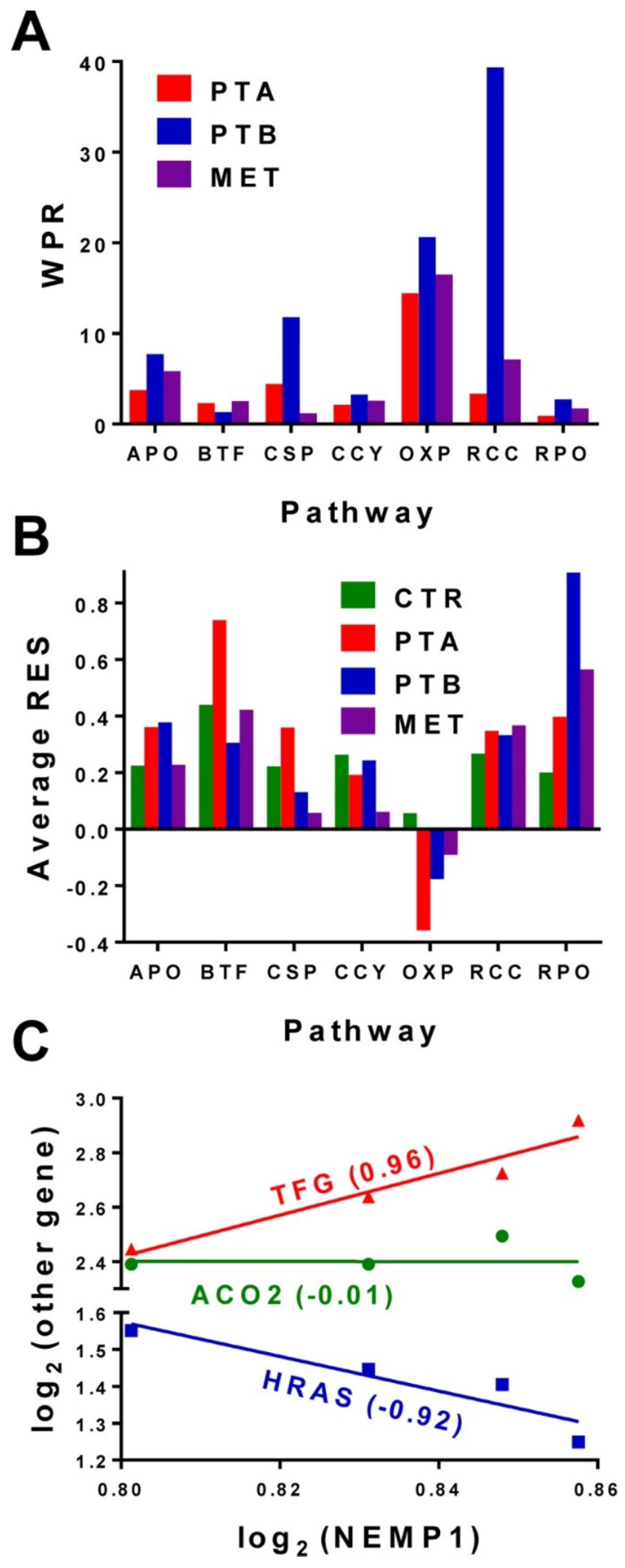
3.2. Expression Stability, Expression Correlation and Weighted Pathway Regulation
3.3. Cancer Nuclei and Surrounding Normal Tissue Are Governed by Distinct GMRs
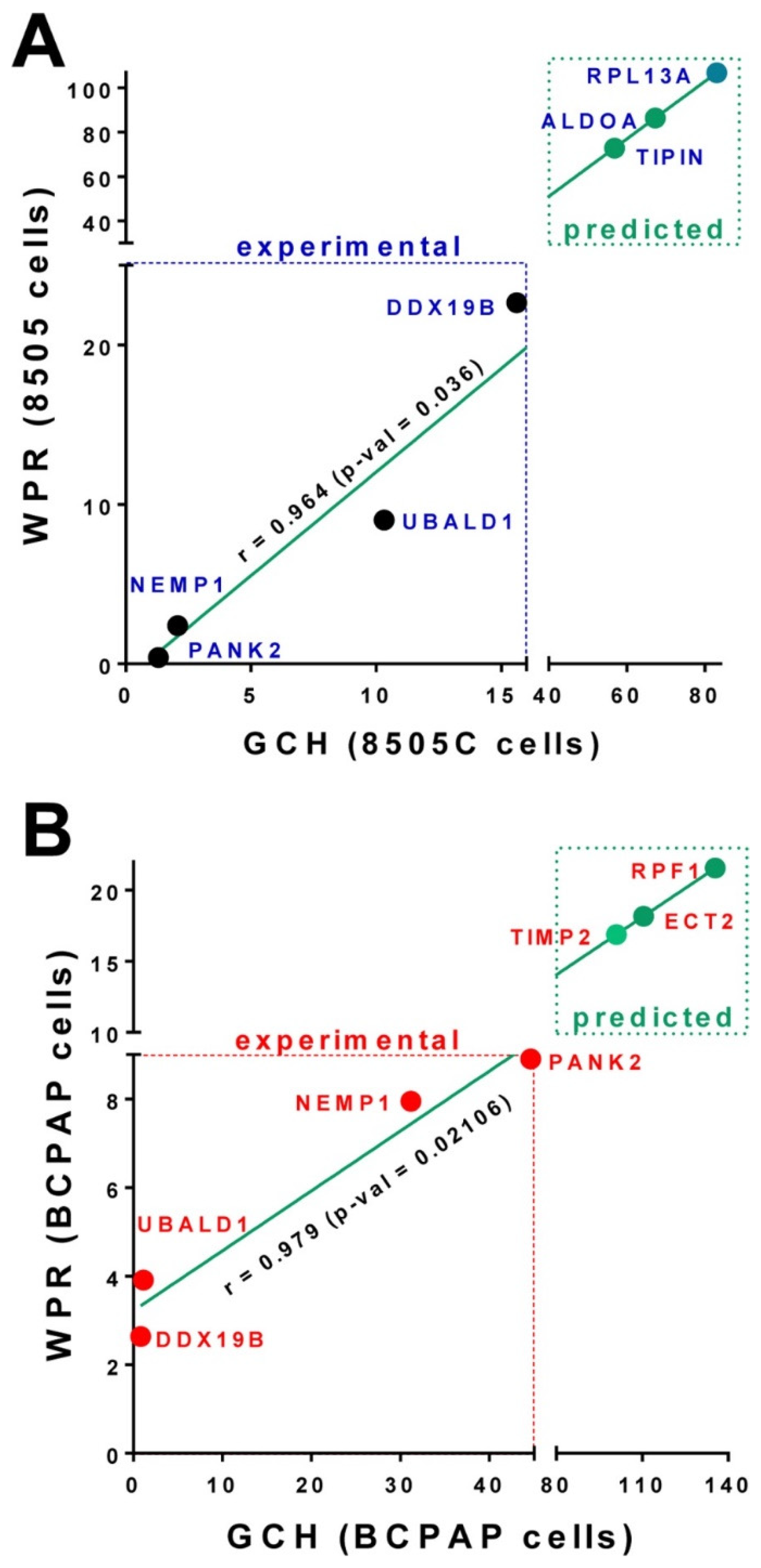
3.4. Experimental Validation of the GMR Theory
3.5. Predicted Transcriptomic Alteration by GMR Manipulation
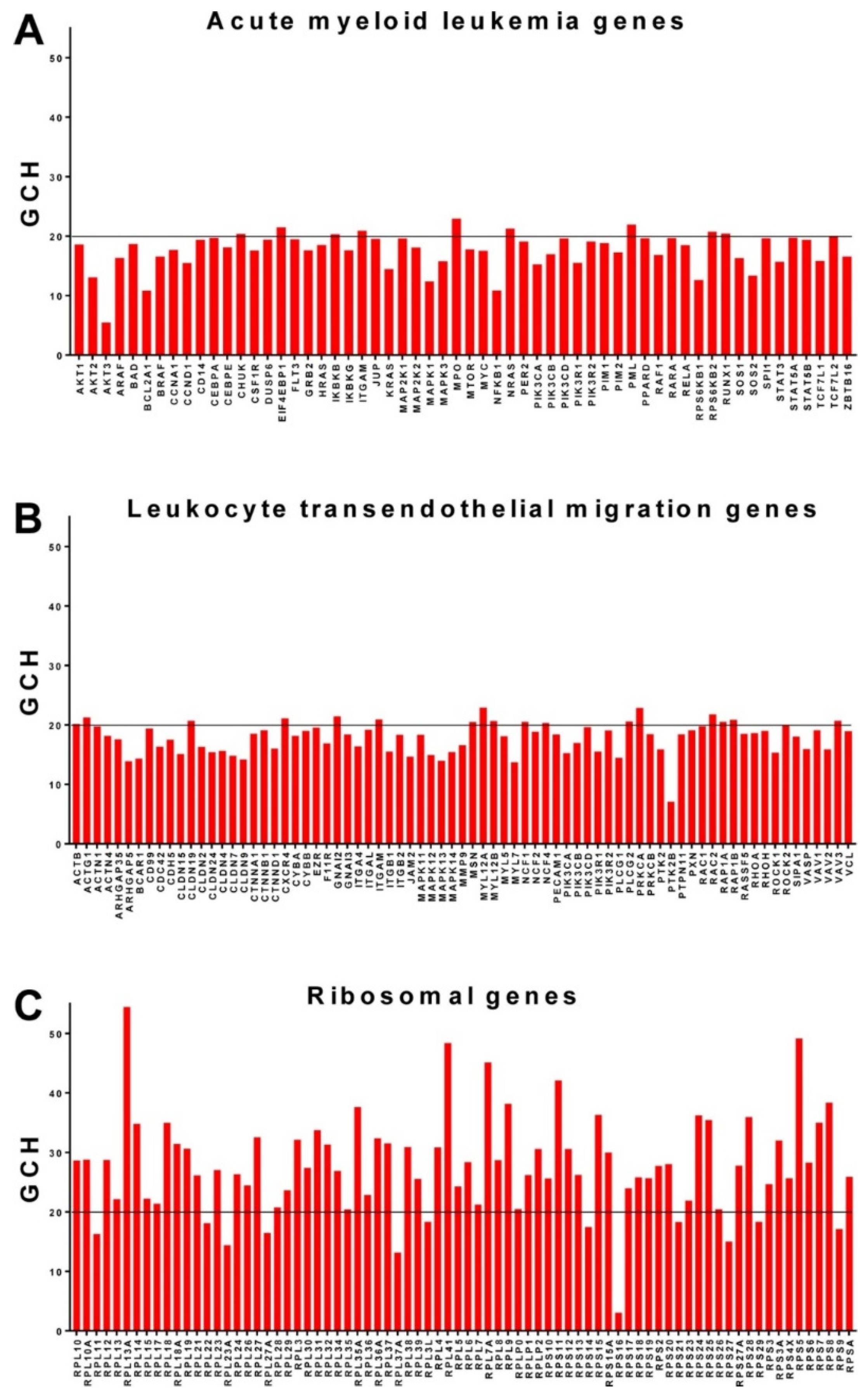
3.6. Ribosomal Genes Top the Hierarchy in the Acute Promyelocytic Leukemia HL-60 Cell Line
4. Discussions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Erstad, D.J.; Fuchs, B.C.; Tanabe, K.K. Molecular signatures in hepatocellular carcinoma: A step toward rationally designed cancer therapy. Cancer 2018, 124, 3084–3104. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, A.; Tilki, D. Biomarkers in prostate cancer—Current clinical utility and future perspectives. Crit. Rev. Oncol. Hematol. 2017, 120, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.; Roszik, J.; Kanikarla-Marie, P.; Davis, J.S.; Morris, J.; Kopetz, S.; Menter, D.G. The potential role of platelets in the consensus molecular subtypes of colorectal cancer. Cancer Metastasis Rev. 2017, 36, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Sacco, A.; Fenotti, A.; Affò, L.; Bazzana, S.; Russo, D.; Presta, M.; Malagola, M.; Anastasia, A.; Motta, M.; Patterson, C.J.; et al. The importance of the genomic landscape in Waldenström’s Macroglobulinemia for targeted therapeutical interventions. Oncotarget 2017, 8, 35435–35444. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Santosh, A.B.; Jones, T.; Harvey, J. A review on oral cancer biomarkers: Understanding the past and learning from the present. J. Cancer Res. Ther. 2016, 12, 486–492. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas (TCGA) Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Mills Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Iacobas, S.; Iacobas, D.A.; Spray, D.C.; Scemes, E. The connexin43 transcriptome during brain development: Importance of genetic background. Brain Res. 2012, 1487, 131–139. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Thomas, N.; Spray, D.C. Sex-dependent gene regulatory networks of the heart rhythm. Funct. Integr. Genomics 2010, 10, 73–86. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Fan, C.; Iacobas, S.; Spray, D.C.; Haddad, G.G. Transcriptomic changes in developing kidney exposed to chronic hypoxia. Biochem. Biophys. Res. Comm. 2006, 349, 329–338. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Fan, C.; Iacobas, S.; Haddad, G.G. Integrated transcriptomic response to cardiac chronic hypoxia: Translation regulators and response to stress in cell survival. Funct. Integr. Genomics 2008, 8, 265–275. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kobets, T.; Iatropoulos, M.J.; Duan, J.D.; Brunnemann, K.D.; Iacobas, D.A.; Iacobas, S.; Vock, E.; Deschl, U.; Williams, G.M. Effects of Nitrosamines on the Expression of Genes Involved in Xenobiotic Metabolism in the Chicken Egg Alternative Genotoxicity Model. Toxicol. Sci. 2018, 166, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, D.A.; Chachua, T.; Iacobas, S.; Benson, M.J.; Borges, K.; Veliskova, J.; Velisek, L. ACTH and PMX53 recover the normal synaptic transcriptome in a rat model of infantile spasms. Sci. Rep. 2018. [Google Scholar] [CrossRef]
- Teng, H.; Mao, F.; Liang, J.; Xue, M.; Wei, W.; Li, X.; Zhang, K.; Feng, D.; Liu, B.; Sun, Z. Transcriptomic signature associated with carcinogenesis and aggressiveness of papillary thyroid carcinoma. Theranostics 2018, 8, 4345–4358. [Google Scholar] [CrossRef] [PubMed]
- Coghlin, C.; Murray, G.I. Biomarkers of colorectal cancer: Recent advances and future challenges. Proteomics Clin. Appl. 2015, 9, 64–71. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Estevez-Garcia, P.; Rivera, F.; Molina-Pinelo, S.; Benavent, M.; Gómez, J.; Limón, M.L.; Pastor, M.D.; Martinez-Perez, J.; Paz-Ares, L.; Carnero, A.; et al. Gene expression profile predictive of response to chemotherapy in metastatic colorectal cancer. Oncotarget 2015, 6, 6151–6159. [Google Scholar] [CrossRef]
- Li, H.; Samawi, H.; Heng, D.Y. The use of prognostic factors in metastatic renal cell carcinoma. Urol. Oncol 2015, 33, 509–516. [Google Scholar] [CrossRef]
- Martínez-Bosch, N.; Rodriguez-Vida, A.; Juanpere, N.; Lloreta, J.; Rovira, A.; Albanell, J.; Bellmunt, J.; Navarro, P. Galectins in prostate and bladder cancer: Tumorigenic roles and clinical opportunities. Nat. Rev. Urol. 2019, 16, 433–445. [Google Scholar] [CrossRef]
- Tang, K.; Xu, H. Prognostic value of meta-signature miRNAs in renal cell carcinoma: An integrated miRNA expression profiling analysis. Sci. Rep. 2015, 14, 10272. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Iacobas, S. Towards a personalized cancer gene therapy: A case of clear cell renal cell carcinoma. Cancer Oncol. Res. 2017, 5. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Tuli, N.; Iacobas, S.; Rasamny, J.K.; Moscatello, A.; Geliebter, J.; Tiwari, R.J. Gene master regulators of papillary and anaplastic thyroid cancer phenotypes. Oncotarget 2018, 9, 2410–2424. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.C.; Owen, M.J. Towards a molecular understanding of T-cell differentiation. Immunol. Today 1991, 12, 86–92. [Google Scholar] [CrossRef][Green Version]
- Doevendans, P.A.; van Bilsen, M. Transcription factors and the cardiac gene programme. Int. J. Biochem. Cell Biol. 1996, 28, 387–403. [Google Scholar] [CrossRef]
- Liyanarachchi, S.; Li, W.; Yan, P.; Bundschuh, R.; Brock, P.; Senter, L.; Ringel, M.D.; de la Chapelle, A.; He, H. Genome-Wide Expression Screening Discloses Long Noncoding RNAs Involved in Thyroid Carcinogenesis. J. Clin. Endocrinol. Metab. 2016, 101, 4005–4013. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Lin, X.D.; Fu, X.H.; Yan, W.; Lin, F.S.; Kuang, P.H.; Luo, Y.; Lin, E.; Hong, X.; Wu, G. Long non-coding RNA BANCR regulates cancer stem cell markers in papillary thyroid cancer via the RAF/MEK/ERK signaling pathway. Oncol. Rep. 2018, 40, 859–866. [Google Scholar] [CrossRef]
- Braicu, C.; Zimta, A.A.; Harangus, A.; Iurca, I.; Irimie, A.; Coza, O.; Berindan-Neagoe, I. The Function of Non-Coding RNAs in Lung Cancer Tumorigenesis. Cancers 2019, 11, 605. [Google Scholar] [CrossRef]
- Braicu, C.; Gulei, D.; Cojocneanu, R.; Raduly, L.; Jurj, A.; Knutsen, E.; Knutsen, E.; Calin, G.A.; Berindan-Neagoe, I. miR-181a/b therapy in lung cancer: Reality or myth? Mol. Oncol. 2019, 13, 9–25. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Haddad, G.G. Heart rhythm genomic fabric in hypoxia. Biochem. Biophys. Res. Commun. 2010, 391, 1769–1774. [Google Scholar] [CrossRef]
- Iacobas, D.A. The Genomic Fabric Perspective on the transcriptome between universal quantifiers and personalized genomic medicine. Biol. Theory 2016, 11, 123–137. [Google Scholar] [CrossRef]
- Gallagher, R.; Collins, S.; Trujillo, J.; McCredie, K.; Ahearn, M.; Tsai, S.; Metzgar, R.; Aulakh, G.; Ting, R.; Ruscetti, F.; et al. Characterization of the continuous, differentiating myeloid cell line (HL-60) from a patient with acute promyelocytic leukemia. Blood 1979, 54, 713–733. [Google Scholar] [PubMed]
- Corso, C.; Ulucan, H.; Parry, E.M.; Parry, J.M. Comparative analysis of two thyroid tumor cell lines by fluorescence in situ hybridization and comparative genomic hybridization. Cancer Genet. Cytogenet. 2002, 137, 108–118. [Google Scholar] [CrossRef]
- Ito, T.; Seyama, T.; Hayashi, Y.; Hayashi, T.; Dohi, K.; Mizuno, T.; Iwamoto, K.; Tsuyama, N.; Nakamura, N.; Akiyama, M. Establishment of 2 human thyroid-carcinoma cell-lines (8305c, 8505c) bearing p53 gene-mutations. Int. J. Oncol. 1994, 4, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Horoszewicz, J.S.; Leong, S.S.; Chu, T.M.; Wajsman, Z.L.; Friedman, M.; Papsidero, L.; Kim, U.; Chai, L.S.; Kakati, S.; Arya, S.K. The LNCaP cell line—A new model for studies on human prostatic carcinoma. Prog. Clin. Biol. Res. 1980, 37, 115–132. [Google Scholar] [PubMed]
- Stone, K.R.; Mickey, D.D.; Wunderli, H.; Mickey, G.H.; Paulson, D.F. Isolation of a human prostate carcinoma cell line (DU 145). Int. J. Cancer 1978, 21, 274–281. [Google Scholar] [CrossRef]
- Kravchick, D.O.; Hrdinka, M.; Iacobas, S.; Iacobas, D.A.; Kreutz, M.R.; Jordan, B.A. Synaptonuclear messenger PRR7 inhibits c-Jun ubiquitination and regulates NMDA mediated excitotoxicity. EMBOJ 2016, 35, 1923–1934. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Urban-Maldonado, M.; Spray, D.C. Sensitivity of the brain transcriptome to connexin ablation. Biochim. Biophys. Acta 2005, 1711, 183–196. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Tanowitz, H.B.; deCarvalho, A.C.; Spray, D.C. Functional genomic fabrics are remodeled in a mouse model of Chagasic cardiomyopathy and restored following cell therapy. Microbes Infect. 2018, 20, 185–195. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Nebieridze, N.; Velisek, L.; Veliskova, J. Estrogen protects neurotransmission transcriptome during status epilepticus. Front Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Spray, D.C. Connexin43 and the brain transcriptome of the newborn mice. Genomics 2007, 89, 113–123. [Google Scholar] [CrossRef][Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Huynh, T.; Xu, S. Gene Annotation Easy Viewer (GAEV): Integrating KEGG’s Gene Function Annotations and Associated Molecular Pathways. F1000Research 2018. [Google Scholar] [CrossRef]
- Dou, R.; Zhang, L.; Lu, T.; Liu, D.; Mei, F.; Huang, J.; Qian, L. Identification of a novel HRAS variant and its association with papillary thyroid carcinoma. Oncol. Lett. 2018, 15, 4511–4516. [Google Scholar] [CrossRef]
- Cipriani, N.A.; Agarwal, S.; Dias-Santagata, D.; Faquin, W.C.; Sadow, P.M. Clear Cell Change in Thyroid Carcinoma: A Clinicopathologic and Molecular Study with Identification of Variable Genetic Anomalies. Thyroid 2017, 27, 819–824. [Google Scholar] [CrossRef]
- Liu, Y.; Tong, C.; Cao, J.; Xiong, M. NEMP1 Promotes Tamoxifen Resistance in Breast Cancer Cells. Biochem. Genet. 2019. [Google Scholar] [CrossRef]
- De Antonellis, P.; Carotenuto, M.; Vandenbussche, J.; De Vita, G.; Ferrucci, V.; Medaglia, C.; Boffa, I.; Galiero, A.; Di Somma, S.; Magliulo, D.; et al. Early targets of miR-34a in neuroblastoma. Mol. Cell Proteomics 2014, 13, 2114–2131. [Google Scholar] [CrossRef]
- Pereira, M.S.; de Almeida, G.C.; Pinto, F.; Viana-Pereira, M.; Reis, R.M. SPINT2 Deregulation in Prostate Carcinoma. J. Histochem. Cytochem. 2016, 64, 32–41. [Google Scholar] [CrossRef]
- Liu, F.; Cox, C.D.; Chowdhury, R.; Dovek, L.; Nguyen, H.; Li, T.; Li, S.; Ozer, B.; Chou, A.; Nguyen, N.; et al. SPINT2 is hypermethylated in both IDH1 mutated and wild-type glioblastomas, and exerts tumor suppression via reduction of c-Met activation. J. Neurooncol. 2019, 142, 423–434. [Google Scholar] [CrossRef]
- Wu, W.F.; Maneix, L.; Insunza, J.; Nalvarte, I.; Antonson, P.; Kere, J.; Yu, N.Y.; Tohonen, V.; Katayama, S.; Einarsdottir, E.; et al. Estrogen receptor β, a regulator of androgen receptor signaling in the mouse ventral prostate. Proc. Natl. Acad. Sci. USA 2017, 114, E3816–E3822. [Google Scholar] [CrossRef]
- Wang, X.F.; Chen, J.; Gong, Y.B.; Qin, Y.C.; Wang, L.; Li, N.C. Long non-coding RNA DUXAP10 promotes the proliferation, migration, and inhibits apoptosis of prostate cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3699–3708. [Google Scholar] [CrossRef]
- Bian, Z.; Yu, Y.; Quan, C.; Guan, R.; Jin, Y.; Wu, J.; Xu, L.; Chen, F.; Bai, J.; Sun, W.; et al. RPL13A as a reference gene for normalizing mRNA transcription of ovarian cancer cells with paclitaxel and 10-hydroxycamptothecin treatments. Mol. Med. Rep. 2015, 11, 3188–3194. [Google Scholar] [CrossRef]
- De Thé, H.; Lavau, C.; Marchio, A.; Chomienne, C.; Degos, L.; Dejean, A. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell 1991, 66, 675–684. [Google Scholar]
- Zelent, A.; Guidez, F.; Melnick, A.; Waxman, S.; Licht, J.D. Translocations of the RARalpha gene in acute promyelocytic leukemia. Oncogene 2001, 20, 7186–7203. [Google Scholar] [CrossRef]
- Zia, M.T.K.; Vinukonda, G.; Vose, L.; Bhimavarapu, B.B.R.; Iacobas, S.; Pandey, N.K.; Beall, A.M.; Dohare, P.; LaGamma, E.F.; Iacobas, D.A.; et al. Postnatal glucocorticoid-induced hypomyelination, gliosis, neurologic deficits are dose-dependent, preparation-specific, and reversible. Exp. Neurol. 2014, 263, 200–213. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENE | DESCRIPTION | CHR | CTR | PTA | PTB | MET |
|---|---|---|---|---|---|---|
| DAPK3 | death-associated protein kinase 3 | 19 | 30.31 | 4.73 | 1.15 | 2.52 |
| PMPCA | peptidase (mitochondrial processing) alpha | 9 | 28.35 | 6.82 | 3.24 | 4.26 |
| COA1 | cytochrome c oxidase assembly factor 1 homolog | 7 | 22.40 | 4.83 | 3.94 | 1.42 |
| FAM208A | family with sequence similarity 208, member A | 3 | 3.08 | 63.97 | 1.59 | 5.40 |
| BCR | breakpoint cluster region | 22 | 1.15 | 57.43 | 1.14 | 1.22 |
| C2orf81 | chromosome 2 open reading frame 81 | 2 | 2.24 | 51.24 | 3.19 | 1.84 |
| FAM27C | family with sequence similarity 27, member C | 9 | 1.75 | 6.03 | 57.19 | 3.73 |
| GTPBP3 | GTP binding protein 3 (mitochondrial) | 19 | 2.07 | 29.80 | 40.06 | 14.01 |
| MIR1915HG | MIR1915 host gene | 10 | 2.57 | 5.55 | 31.14 | 4.06 |
| ALG13 | ALG13, UDP-N-acetylglucosaminyltransferase subunit | X | 3.64 | 9.97 | 2.12 | 82.95 |
| NUDT18 | nudix (nucleoside diphosphate linked moiety X)-type motif 18 | 8 | 1.64 | 2.69 | 1.89 | 48.40 |
| RAD54B | RAD54 homolog B | 8 | 0.96 | 6.10 | 4.09 | 40.02 |
| GENE | DESCRIPTION | CHR | NORM | PAP-C | BCPAP | 8505C |
|---|---|---|---|---|---|---|
| RASD1 | RAS, dexamethasone-induced 1 | 17 | 41.51 | 4.50 | 5.70 | 7.31 |
| POTEF | POTE ankyrin domain family, member F | 2 | 31.17 | 8.50 | 6.90 | 6.36 |
| RCN2 | reticulocalbin 2, EF-hand calcium binding domain | 15 | 31.09 | 5.53 | 7.99 | 10.38 |
| SPINT2 | serine peptidase inhibitor, Kunitz type, 2 | 19 | 1.93 | 54.97 | 18.83 | 5.88 |
| RPAP3 | RNA polymerase II associated protein 3 | 12 | 5.33 | 51.74 | 3.25 | 12.69 |
| BZW1 | basic leucine zipper and W2 domains 1 | 2 | 2.67 | 44.32 | 12.77 | 26.73 |
| RPF1 | ribosome production factor 1 homolog | 1 | 8.36 | 2.22 | 135.50 | 22.11 |
| TIMP2 | TIMP metallopeptidase inhibitor 2 | 17 | 2.68 | 6.36 | 110.45 | 18.04 |
| ECT2 | epithelial cell transforming 2 | 3 | 6.93 | 8.16 | 100.98 | 28.15 |
| SENP5 | SUMO1/sentrin specific peptidase 5 | 3 | 9.93 | 6.32 | 100.37 | 13.71 |
| RPL13A | ribosomal protein L13a | 19 | 13.16 | 8.73 | 63.26 | 83.02 |
| ALDOA | aldolase A, fructose-bisphosphate | 16 | 7.00 | 28.05 | 2.59 | 67.30 |
| TIPIN | TIMELESS interacting protein | 15 | 3.11 | 9.15 | 37.04 | 56.85 |
| GENE | DESCRIPTION | CHR | NORM1 | CANCER1 | NORM2 | CANCER2 | LNCaP | DU145 |
|---|---|---|---|---|---|---|---|---|
| TOR1A | torsin family 1, member A | 9 | 84.24 | 1.91 | 3.27 | 10.94 | 6.57 | 16.47 |
| MRPS12 | mitochondrial ribosomal protein S12 | 19 | 80.71 | 4.09 | 3.50 | 3.04 | 12.16 | 15.71 |
| GTF2H1 | general transcription factor IIH, polypeptide 1 | 11 | 42.66 | 5.71 | 5.27 | 5.83 | 4.34 | 17.34 |
| BAIAP2L1 | BAI1-associated protein 2-like 1 | 7 | 2.06 | 49.38 | 0.86 | 2.56 | 3.72 | 15.95 |
| FAM71E1 | family with sequence similarity 71, member E1 | 19 | 0.93 | 48.21 | 1.08 | 4.49 | 3.59 | 16.26 |
| MAP6D1 | MAP6 domain containing 1 | 3 | 1.29 | 45.26 | 1.34 | 2.05 | 7.50 | 16.61 |
| SFR1 | SWI5-dependent recombination repair 1 | 10 | 2.66 | 1.36 | 40.10 | 4.64 | 5.01 | 17.10 |
| EDF1 | endothelial differentiation-related factor 1 | 9 | 2.52 | 1.86 | 29.51 | 5.75 | 5.18 | 17.25 |
| RHOD | ras homolog family member D | 11 | 1.25 | 1.15 | 27.90 | 2.50 | 3.92 | 14.89 |
| LOC145474 | uncharacterized long non-coding RNA | 14 | 2.23 | 1.64 | 1.16 | 126.75 | 1.01 | 12.33 |
| PRRG1 | proline rich Gla (G-carboxyglutamic acid) 1 | X | N/A | N/A | 1.82 | 87.53 | 5.10 | 14.33 |
| ASAP3 | ArfGAP with SH3 domain, ankyrin repeat and PH domain 3 | 1 | 1.19 | 1.73 | 1.97 | 76.23 | 4.15 | 16.28 |
| WFDC3 | WAP four-disulfide core domain 3 | 20 | 3.57 | 1.90 | 1.33 | 11.61 | 173.58 | 15.89 |
| RPL31 | 60S ribosomal protein L31 | 2 | 1.30 | 0.94 | 2.19 | 8.25 | 39.11 | 18.16 |
| ALX4 | ALX homeobox 4 | 11 | N/A | N/A | N/A | 6.32 | 35.18 | 18.16 |
| VIM | vimentin | 10 | 1.28 | 1.97 | N/A | 2.85 | 3.51 | 33.95 |
| POTEM | POTE ankyrin domain family, member M | 14 | 1.18 | 2.40 | 0.54 | 4.18 | 2.51 | 33.25 |
| EXOC5 | exocyst complex component 5 | 14 | 1.32 | 1.32 | 1.03 | 5.28 | 2.75 | 32.19 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iacobas, S.; Ede, N.; Iacobas, D.A. The Gene Master Regulators (GMR) Approach Provides Legitimate Targets for Personalized, Time-Sensitive Cancer Gene Therapy. Genes 2019, 10, 560. https://doi.org/10.3390/genes10080560
Iacobas S, Ede N, Iacobas DA. The Gene Master Regulators (GMR) Approach Provides Legitimate Targets for Personalized, Time-Sensitive Cancer Gene Therapy. Genes. 2019; 10(8):560. https://doi.org/10.3390/genes10080560
Chicago/Turabian StyleIacobas, Sanda, Nneka Ede, and Dumitru A. Iacobas. 2019. "The Gene Master Regulators (GMR) Approach Provides Legitimate Targets for Personalized, Time-Sensitive Cancer Gene Therapy" Genes 10, no. 8: 560. https://doi.org/10.3390/genes10080560
APA StyleIacobas, S., Ede, N., & Iacobas, D. A. (2019). The Gene Master Regulators (GMR) Approach Provides Legitimate Targets for Personalized, Time-Sensitive Cancer Gene Therapy. Genes, 10(8), 560. https://doi.org/10.3390/genes10080560