De Novo Transcriptome Assembly and Annotation of Liver and Brain Tissues of Common Brushtail Possums (Trichosurus vulpecula) in New Zealand: Transcriptome Diversity after Decades of Population Control

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Ethics Statement
2.2. Specimen Collection
2.3. Nucleic Acid Extraction, Genomic Library Preparation, and Sequencing
2.4. Transcriptome Assembly, Variant Calling and Functional Annotation
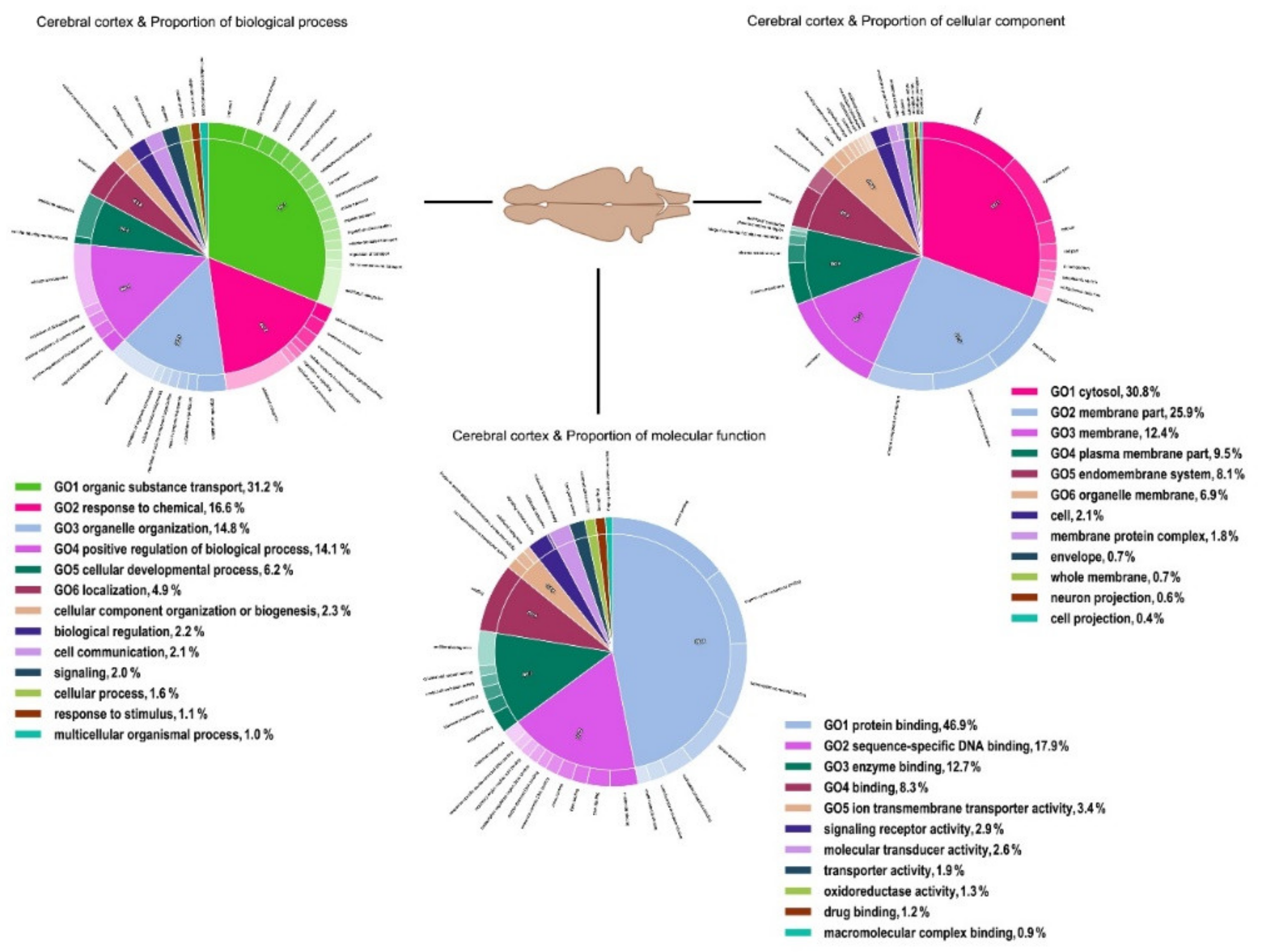
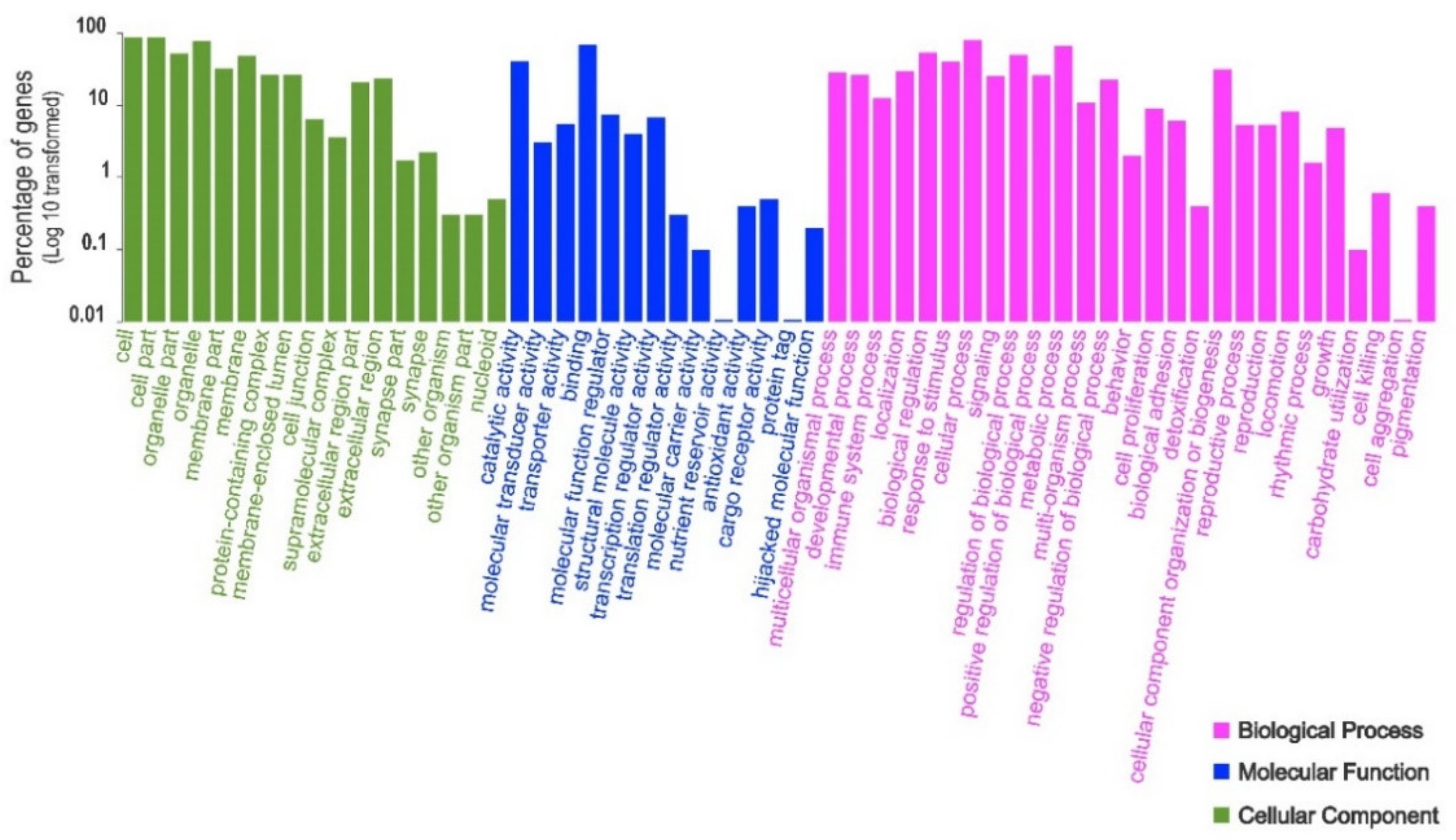
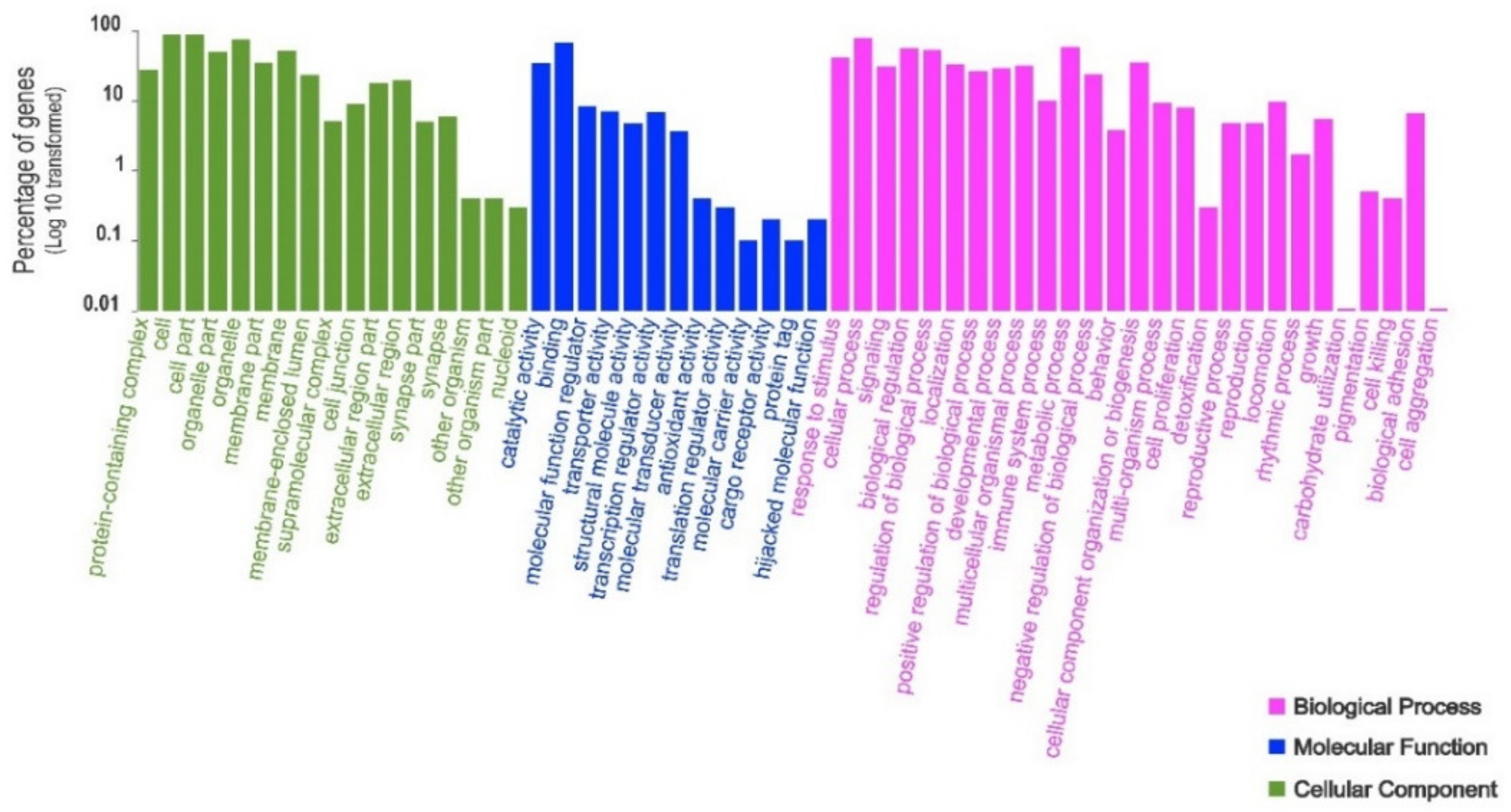
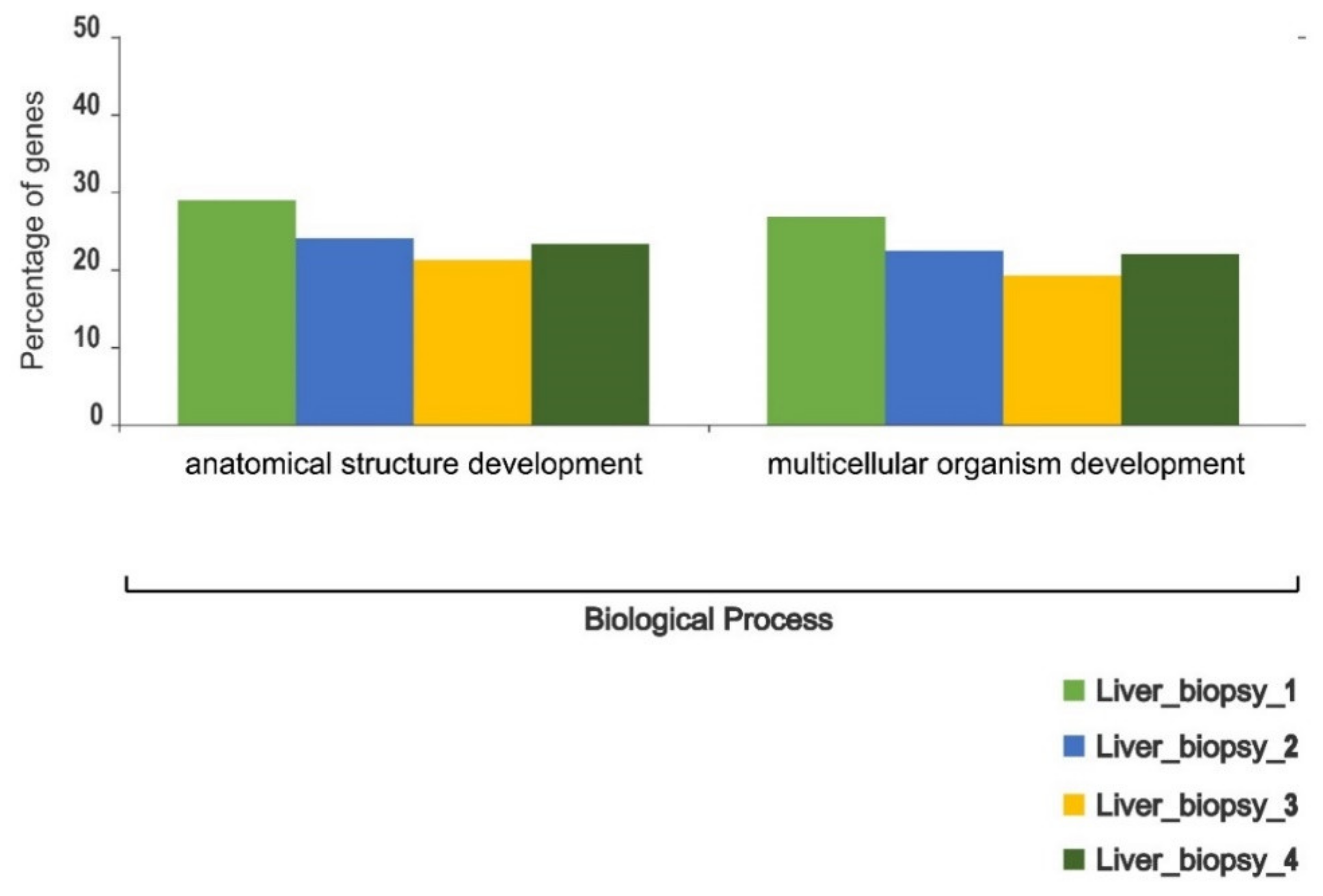
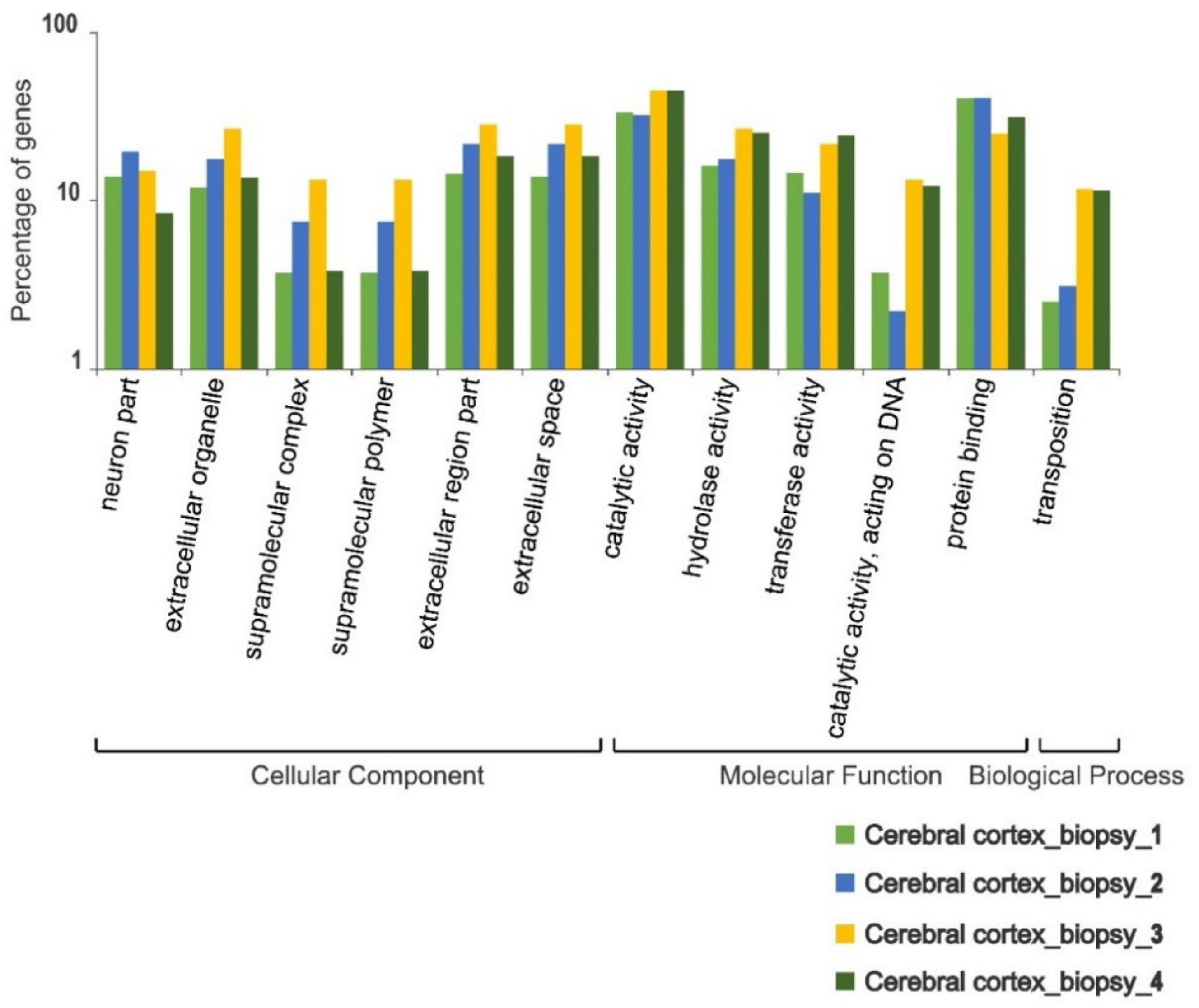
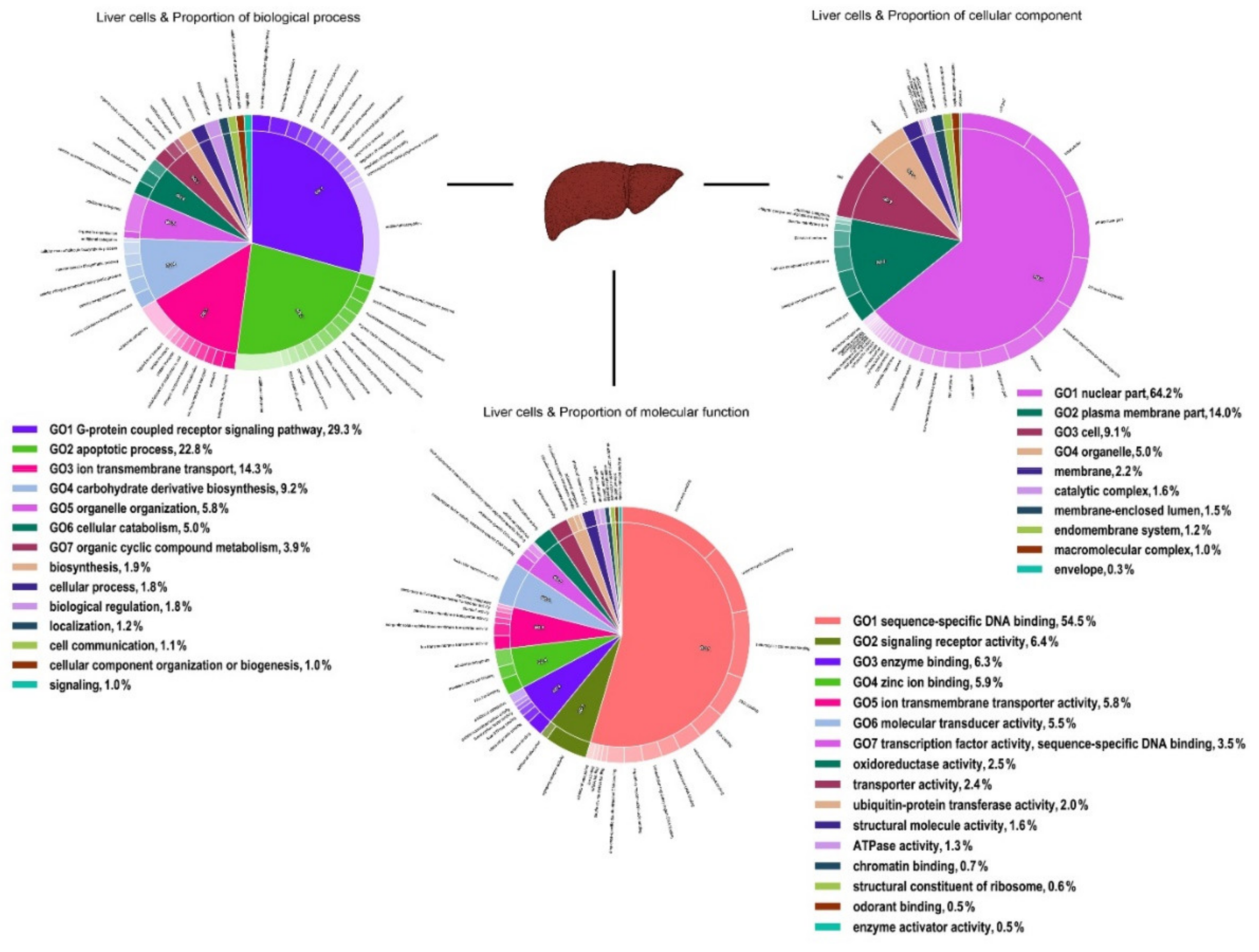
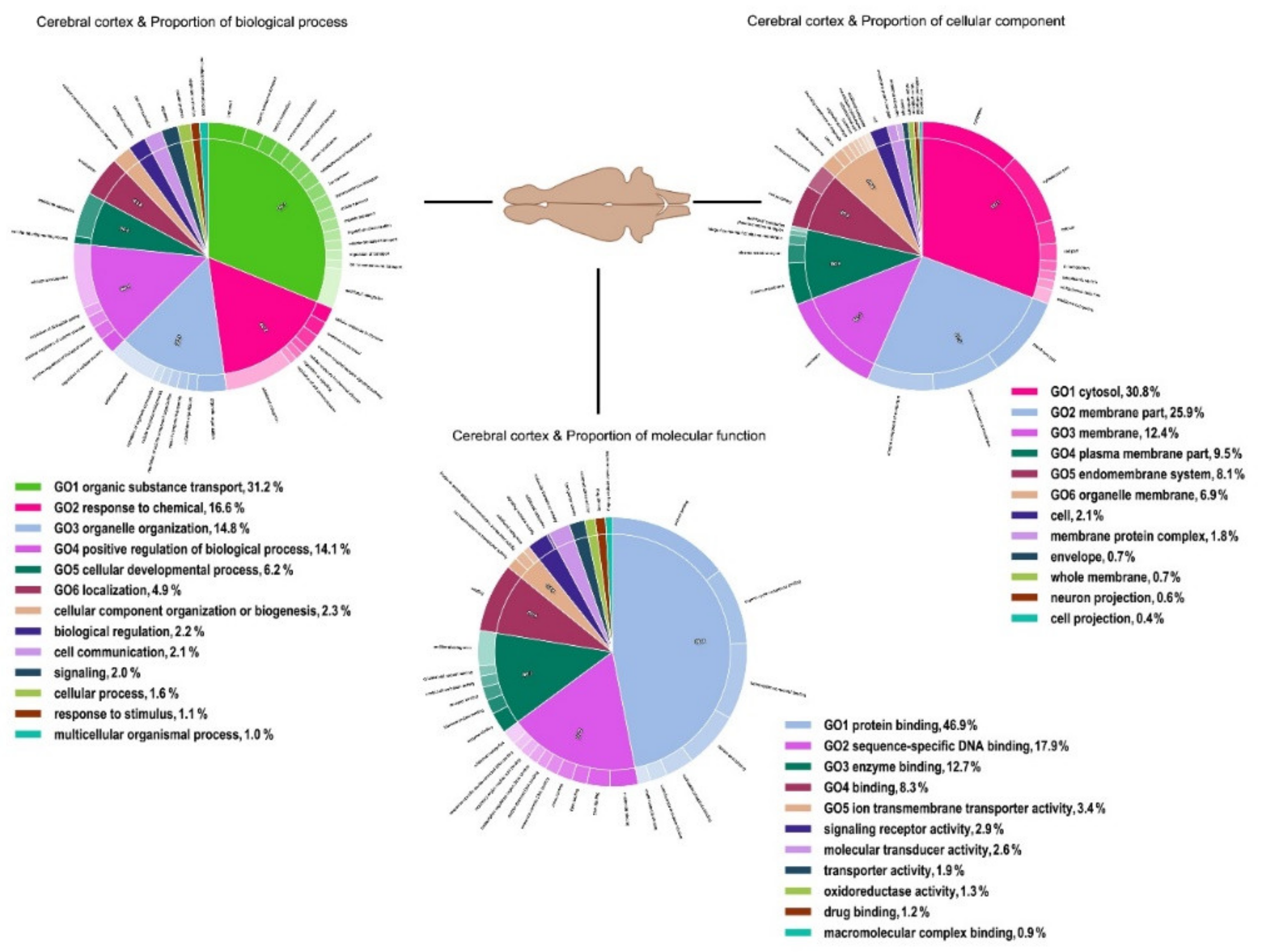
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Laird, M.; Bradshaw, J. The Break-up of a Long-Term Relationship: The Cretaceous Separation of New Zealand from Gondwana. Gondwana Res. 2004, 7, 273–286. [Google Scholar] [CrossRef]
- Wilmshurst, J.M.; Anderson, A.J.; Higham, T.F.; Worthy, T.H. Dating the Late Prehistoric Dispersal of Polynesians to New Zealand Using the Commensal Pacific Rat. Proc. Natl. Acad. Sci. USA 2008, 105, 7676–7680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilmshurst, J.M.; Higham, T.F.; Allen, H.; Johns, D.; Phillips, C. Early Maori Settlement Impacts in Northern Coastal Taranaki, New Zealand. N. Zl. J. Ecol. 2004, 28, 167–179. [Google Scholar]
- Parkes, J.; Murphy, E. Management of Introduced Mammals in New Zealand. N. Z J. Zool. 2003, 30, 335–359. [Google Scholar] [CrossRef] [Green Version]
- Robertson, H.; Baird, K.; Dowding, J.; Elliott, G.; Hitchmough, R.; Miskelly, C.; McArthur, N.; O’Donnell, C.; Sagar, P.; Scofield, R. Conservation Status of New Zealand Birds, 2016; New Zealand Threat Classification Series 19; New Zealand Department of Conservation: Wellington, New Zealand, 2017. [Google Scholar]
- Emami-Khoyi, A.; Paterson, A.M.; Hartley, D.A.; Boren, L.J.; Cruickshank, R.H.; Ross, J.G.; Murphy, E.C.; Else, T.-A. Mitogenomics Data Reveal Effective Population Size, Historical Bottlenecks, and the Effects of Hunting on New Zealand Fur Seals (Arctocephalus Forsteri). Mitochondrial DNA Part A 2018, 29, 567–580. [Google Scholar] [CrossRef]
- Thomson, G.M. The Naturalisation of Animals and Plants in New Zealand; Cambridge University Press: Cambridge, UK, 1922. [Google Scholar]
- Pracy, L.T. Introduction and Liberation of the Opossum (Trichosurus vulpecula) into New Zealand; New Zealand Forest Service: New Zealand, 1962. [Google Scholar]
- Cowan, P. Brushtail Possum. In The Handbook of New Zealand Mammals, 2nd ed.; Oxford University Press: Oxford, UK, 2005. [Google Scholar]
- Brown, K.; Innes, J.; Shorten, R. Evidence That Possums Prey on and Scavenge Birds’ Eggs, Birds and Mammals. Notornis 1993, 40, 169–177. [Google Scholar]
- Murphy, E.C.; Russell, J.C.; Broome, K.G.; Ryan, G.J.; Dowding, J.E. Conserving New Zealand’s Native Fauna: A Review of Tools Being Developed for the Predator Free 2050 Programme. J. Ornithol. 2019, 160, 883–892. [Google Scholar] [CrossRef]
- Green, W. The Use of 1080 for Pest Control: A Discussion Document; Animal Health Board and the Department of Conservation: New Zealand, 2004. [Google Scholar]
- Ogilvie, S.; Miller, A.; Ataria, J.M.; Waiwai, J.; Doherty, J. Uptake of 1080 by Watercress and Puha—Culturally Important Plants Used for Food; Lincoln University Wildlife Management Report series [59]; Lincoln University: Christchurch, New Zealand, 2009. [Google Scholar]
- Eason, C.; Miller, A.; Ogilvie, S.; Fairweather, A. An Updated Review of the Toxicology and Ecotoxicology of Sodium Fluoroacetate (1080) in Relation to Its Use as a Pest Control Tool in New Zealand. N. Zeal. J. Ecol. 2011, 35, 1–20. [Google Scholar]
- King, D.; Oliver, A.; Mead, R. The Adaptation of Some Western Australian Mammals to Food Plants Containing Fluoroacetate. Aust. J. Zool. 1978, 26, 699–712. [Google Scholar] [CrossRef]
- Prentis, P.J.; Wilson, J.R.; Dormontt, E.E.; Richardson, D.M.; Lowe, A.J. Adaptive Evolution in Invasive Species. Trends Plant Sci. 2008, 13, 288–294. [Google Scholar] [CrossRef]
- Leger, E.A.; Rice, K.J. Invasive California Poppies (Eschscholzia Californica) Grow Larger Than Native Individuals under Reduced Competition. Ecol. Lett. 2003, 6, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Triggs, S.; Green, W. Geographic Patterns of Genetic Variation in Brushtail Possums Trichosurus vulpecula and Implications for Pest Control. N. Zeal. J. Ecol. 1989, 12, 1–10. [Google Scholar]
- Sarre, S.D.; Aitken, N.; Adamack, A.T.; MacDonald, A.J.; Gruber, B.; Cowan, P. Creating New Evolutionary Pathways through Bioinvasion: The Population Genetics of Brushtail Possums in New Zealand. Mol. Ecol. 2014, 23, 3419–3433. [Google Scholar] [CrossRef] [PubMed]
- Cao, D. De Novo Transcriptome Analysis of Taxus Chinensis Var. Mairei to Identify Significant Pathways Associated with the Fruit Color of This Species. Biochem. Syst. Ecol. 2019, 84, 1–7. [Google Scholar] [CrossRef]
- Stahl, B.A.; Gross, J.B.; Speiser, D.I.; Oakley, T.H.; Patel, N.H.; Gould, D.B.; Protas, M.E. A Transcriptomic Analysis of Cave, Surface, and Hybrid Isopod Crustaceans of the Species Asellus Aquaticus. PLoS ONE 2015, 10, e0140484. [Google Scholar] [CrossRef] [Green Version]
- Soewarto, J.; Hamelin, C.; Bocs, S.; Mournet, P.; Vignes, H.; Berger, A.; Armero, A.; Martin, G.; Dereeper, A.; Sarah, G. Transcriptome Data from Three Endemic Myrtaceae Species from New Caledonia Displaying Contrasting Responses to Myrtle Rust (Austropuccinia Psidii). Data Brief 2019, 22, 794–811. [Google Scholar] [CrossRef]
- Liu, G.-H.; Xu, M.-J.; Chang, Q.-C.; Gao, J.-F.; Wang, C.-R.; Zhu, X.-Q. De Novo Transcriptomic Analysis of the Female and Male Adults of the Blood Fluke Schistosoma turkestanicum. Parasites Vectors 2016, 9, 143. [Google Scholar] [CrossRef] [Green Version]
- Bankers, L.; Neiman, M. De Novo Transcriptome Characterization of a Sterilizing Trematode Parasite (Microphallus Sp.) from Two Species of New Zealand Snails. G3 Genes Genomes Genet. 2017, 7, 871–880. [Google Scholar]
- Yang, M.; Wang, Q.; Wang, S.; Wang, Y.; Zeng, Q.; Qin, Q. Transcriptomics Analysis Reveals Candidate Genes and Pathways for Susceptibility or Resistance to Singapore Grouper Iridovirus in Orange-Spotted Grouper (Epinephelus coioides). Dev. Comp. Immunol. 2019, 90, 70–79. [Google Scholar] [CrossRef]
- Sarwar, M.B.; Ahmad, Z.; Rashid, B.; Hassan, S.; Gregersen, P.L.; De la O. Leyva, M.; Nagy, I.; Asp, T.; Husnain, T. De Novo Assembly of Agave sisalana Transcriptome in Response to Drought Stress Provides Insight into the Tolerance Mechanisms. Sci. Rep. 2019, 9, 396. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.V.; Bohonak, A.J.; Simovich, M.A.; Goddard, N.S.; Graige, N.S. Discovery and Validation of Species–Specific Diagnostic SNP Markers for the Endangered San Diego Fairy Shrimp (Branchinecta sandiegonensis) and the Versatile Fairy Shrimp (Branchinecta lindahli). Conserv. Genet. Resour. 2018, 10, 897–905. [Google Scholar] [CrossRef]
- Chen, X.; Mei, J.; Wu, J.; Jing, J.; Ma, W.; Zhang, J.; Dan, C.; Wang, W.; Gui, J.-F. A Comprehensive Transcriptome Provides Candidate Genes for Sex Determination/Differentiation and SSR/SNP Markers in Yellow Catfish. Mar. Biotechnol. 2015, 17, 190–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipka, A.; Paukszto, L.; Majewska, M.; Jastrzebski, J.P.; Panasiewicz, G.; Szafranska, B. De Novo Characterization of Placental Transcriptome in the Eurasian Beaver (Castor fiber L.). Funct. Integr. Genom. 2019, 19, 421–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dearden, P.K.; Gemmell, N.J.; Mercier, O.R.; Lester, P.J.; Scott, M.J.; Newcomb, R.D.; Buckley, T.R.; Jacobs, J.M.; Goldson, S.G.; Penman, D.R. The Potential for the Use of Gene Drives for Pest Control in New Zealand: A Perspective. J. R. Soc. N. Zeal. 2018, 48, 225–244. [Google Scholar] [CrossRef]
- Hu, D.G.; Marri, S.; McKinnon, R.A.; Mackenzie, P.I.; Meech, R. Deregulation of the Genes That Are Involved in Drug Absorption, Distribution, Metabolism, and Excretion in Hepatocellular Carcinoma. J. Pharmacol. Exp. Ther. 2019, 368, 363–381. [Google Scholar] [CrossRef] [Green Version]
- Porseryd, T.; Volkova, K.; Reyhanian Caspillo, N.; Källman, T.; Dinnetz, P.; Porsh Hällström, I. Persistent Effects of Developmental Exposure to 17α-Ethinylestradiol on the Zebrafish (Danio rerio) Brain Transcriptome and Behavior. Front. Behav. Neurosci. 2017, 11, 69. [Google Scholar] [CrossRef] [Green Version]
- Saaristo, M.; Wong, B.B.; Mincarelli, L.; Craig, A.; Johnstone, C.P.; Allinson, M.; Lindström, K.; Craft, J.A. Characterisation of the Transcriptome of Male and Female Wild-Type Guppy Brains with RNA-Seq and Consequences of Exposure to the Pharmaceutical Pollutant, 17α-Ethinyl Estradiol. Aquat. Toxicol. 2017, 186, 28–39. [Google Scholar] [CrossRef]
- Tourancheau, A.; Rouleau, M.; Guauque-Olarte, S.; Villeneuve, L.; Gilbert, I.; Droit, A.; Guillemette, C. Quantitative Profiling of the UGT Transcriptome in Human Drug-Metabolizing Tissues. Pharm. J. 2018, 18, 251. [Google Scholar] [CrossRef] [Green Version]
- Zygmunt, M.; Piechota, M.; Rodriguez Parkitna, J.; Korostyński, M. Decoding the Transcriptional Programs Activated by Psychotropic Drugs in the Brain. Genes Brain Behav. 2019, 18, e12511. [Google Scholar] [CrossRef]
- Tolosa, L.; Jiménez, N.; Pelechá, M.; Castell, J.V.; Gómez-Lechón, M.J.; Donato, M.T. Long-Term and Mechanistic Evaluation of Drug-Induced Liver Injury in Upcyte Human Hepatocytes. Arch. Toxicol. 2019, 93, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Kontkanen, O.; Törönen, P.; Lakso, M.; Wong, G.; Castrén, E. Antipsychotic Drug Treatment Induces Differential Gene Expression in the Rat Cortex. J. Neurochem. 2002, 83, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Corton, J.C.; Bushel, P.R.; Fostel, J.; Raegan, B. Sources of Variance in Baseline Gene Expression in the Rodent Liver. Mutat. Res./Genet. Toxicol. Environ. Mutagenesis 2012, 746, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merienne, N.; Meunier, C.; Schneider, A.; Seguin, J.; Nair, S.S.; Rocher, A.B.; Le Gras, S.; Keime, C.; Faull, R.; Pellerin, L. Cell-Type-Specific Gene Expression Profiling in Adult Mouse Bain Reveals Normal and Disease-State Signatures. Cell Rep. 2019, 26, 2477–2493.e2479. [Google Scholar] [CrossRef] [Green Version]
- Nadler, J.J.; Zou, F.; Huang, H.; Moy, S.S.; Lauder, J.; Crawley, J.N.; Threadgill, D.W.; Wright, F.A.; Magnuson, T.R. Large-Scale Gene Expression Differences across Brain Regions and Inbred Strains Correlate with a Behavioral Phenotype. Genetics 2006, 174, 1229–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De Novo Transcript Sequence Reconstruction from RNA-Seq Using the Trinity Platform for Reference Generation and Analysis. Nat. Protoc. 2013, 8, 1494. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.; Papanicolaou, A. Transdecoder (Find Coding Regions within Transcripts). 2016. Available online: https://github.com/TransDecoder/TransDecoder (accessed on 17 June 2019).
- Bryant, D.M.; Johnson, K.; DiTommaso, T.; Tickle, T.; Couger, M.B.; Payzin-Dogru, D.; Lee, T.J.; Leigh, N.D.; Kuo, T.-H.; Davis, F.G. A Tissue-Mapped Axolotl De Novo Transcriptome Enables Identification of Limb Regeneration Factors. Cell Rep. 2017, 18, 762–776. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER Web Server: Interactive Sequence Similarity Searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [Green Version]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting Transmembrane Protein Topology with a Hidden Markov Model: Application to Complete Genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and Rapid Annotation of Ribosomal RNA Genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Seppey, M.; Manni, M.; Zdobnov, E.M. Busco: Assessing Genome Assembly and Annotation Completeness. In Gene Prediction; Springer: Berlin/Heidelberg, Germany, 2019; pp. 227–245. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic Mutation and Copy Number Alteration Discovery in Cancer by Exome Sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Sacomoto, G.A.; Kielbassa, J.; Chikhi, R.; Uricaru, R.; Antoniou, P.; Sagot, M.-F.; Peterlongo, P.; Lacroix, V. KISSPLICE: De-Novo Calling Alternative Splicing Events from Rna-Seq Data. BMC Bioinform. 2012, 13, S5. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from Rna-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25. [Google Scholar] [CrossRef] [Green Version]
- Reimand, J.; Kull, M.; Peterson, H.; Hansen, J.; Vilo, J. G: Profiler—A Web-Based Toolset for Functional Profiling of Gene Lists from Large-Scale Experiments. Nucleic Acids Res. 2007, 35, W193–W200. [Google Scholar] [CrossRef]
- Ye, J.; Zhang, Y.; Cui, H.; Liu, J.; Wu, Y.; Cheng, Y.; Xu, H.; Huang, X.; Li, S.; Zhou, A. WEGO 2.0: A Web Tool for Analyzing and Plotting Go Annotations, 2018 Update. Nucleic Acids Res. 2018, 46, W71–W75. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsova, I.; Lugmayr, A.; Siira, S.J.; Rackham, O.; Filipovska, A. CirGO: An Alternative Circular Way of Visualising Gene Ontology Terms. BMC Bioinform. 2019, 20, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosakovsky Pond, S.L.; Frost, S.D. Not So Different after All: A Comparison of Methods for Detecting Amino Acid Sites under Selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [Green Version]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y. KEGG Mapper for Inferring Cellular Functions from Protein Sequences. Protein Sci. 2019, 29, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Huynh, T.; Xu, S. Gene Annotation Easy Viewer (Gaev): Integrating KEGG’s Gene Function Annotations and Associated Molecular Pathways. F1000Research 2018, 7, 416. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Dunlop, E.S.; McLaughlin, R.; Adams, J.V.; Jones, M.; Birceanu, O.; Christie, M.R.; Criger, L.A.; Hinderer, J.L.; Hollingworth, R.M.; Johnson, N.S. Rapid Evolution Meets Invasive Species Control: The Potential for Pesticide Resistance in Sea Lamprey. Can. J. Fish. Aquat. Sci. 2017, 75, 152–168. [Google Scholar] [CrossRef]
- Tammer, I.; Geginat, G.; Lange, S.; Kropf, S.; Lodes, U.; Schlüter, D.; Lippert, H.; Meyer, F. Antibiotic Consumption and the Development of Antibiotic Resistance in Surgical Units. Zent. Fur Chir. 2016, 141, 53–61. [Google Scholar]
- Vontas, J.; Grigoraki, L.; Morgan, J.; Tsakireli, D.; Fuseini, G.; Segura, L.; de Carvalho, J.N.; Nguema, R.; Weetman, D.; Slotman, M.A. Rapid Selection of a Pyrethroid Metabolic Enzyme CYP9K1 by Operational Malaria Control Activities. Proc. Natl. Acad. Sci. USA 2018, 115, 4619–4624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nacci, D.; Proestou, D.; Champlin, D.; Martinson, J.; Waits, E.R. Genetic Basis for Rapidly Evolved Tolerance in the Wild: Adaptation to Toxic Pollutants by an Estuarine Fish Species. Mol. Ecol. 2016, 25, 5467–5482. [Google Scholar] [CrossRef] [PubMed]
- Twigg, L.E.; Martin, G.R.; Lowe, T.J. Evidence of Pesticide Resistance in Medium-Sized Mammalian Pests: A Case Study with 1080 Poison and Australian Rabbits. J. Appl. Ecol. 2002, 39, 549–560. [Google Scholar] [CrossRef] [Green Version]
- Cowan, P.E.; Gleeson, D.M.; Howitt, R.L.; Ramón-Laca, A.; Esther, A.; Pelz, H.J. Vkorc1 Sequencing Suggests Anticoagulant Resistance in Rats in New Zealand. Pest Manag. Sci. 2017, 73, 262–266. [Google Scholar] [CrossRef]
- Joseph, P. Transcriptomics in Toxicology. Food Chem. Toxicol. 2017, 109, 650–662. [Google Scholar] [CrossRef]
- Kohonen, P.; Parkkinen, J.A.; Willighagen, E.L.; Ceder, R.; Wennerberg, K.; Kaski, S.; Grafström, R.C. A Transcriptomics Data-Driven Gene Space Accurately Predicts Liver Cytopathology and Drug-Induced Liver Injury. Nat. Commun. 2017, 8, 15932. [Google Scholar] [CrossRef]
- Huo, C.; Liu, X.; Zhao, J.; Zhao, T.; Huang, H.; Ye, H. Abnormalities in Behaviour, Histology and Prefrontal Cortical Gene Expression Profiles Relevant to Schizophrenia in Embryonic Day 17 Mam-Exposed C57bl/6 Mice. Neuropharmacology 2018, 140, 287–301. [Google Scholar] [CrossRef]
- Lisowski, P.; Wieczorek, M.; Goscik, J.; Juszczak, G.R.; Stankiewicz, A.M.; Zwierzchowski, L.; Swiergiel, A.H. Effects of Chronic Stress on Prefrontal Cortex Transcriptome in Mice Displaying Different Genetic Backgrounds. J. Mol. Neurosci. 2013, 50, 33–57. [Google Scholar] [CrossRef] [Green Version]
- Guengerich, F.P. Cytochrome P450 and Chemical Toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M.; Sim, S.C.; Gomez, A.; Rodriguez-Antona, C. Influence of Cytochrome P450 Polymorphisms on Drug Therapies: Pharmacogenetic, Pharmacoepigenetic and Clinical Aspects. Pharmacol. Ther. 2007, 116, 496–526. [Google Scholar] [CrossRef]
- Johnson, R.N.; O’Meally, D.; Chen, Z.; Etherington, G.J.; Ho, S.Y.; Nash, W.J.; Grueber, C.E.; Cheng, Y.; Whittington, C.M.; Dennison, S. Adaptation and Conservation Insights from the Koala Genome. Nat. Genet. 2018, 50, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Mead, R.J. The Biochemistry and Co-Evolutionary Role of Monofluoracetic Acid in Relation to Plant-Animal Interaction in Australia; University of Western Australia: Perth, Australia, 1980. [Google Scholar]
- Twigg, L.; Mead, R.; King, D. Metabolism of Fluoroacetate in the Skink (Tiliqua rugosa) and the Rat (Rattus norvegicus). Aust. J. Biol. Sci. 1986, 39, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Twigg, L.E.; King, D.R. The Impact of Fluoroacetate-Bearing Vegetation on Native Australian Fauna: A Review. Oikos 1991, 61, 412–430. [Google Scholar] [CrossRef] [Green Version]
- Zaman, G.; Flens, M.; Van Leusden, M.; De Haas, M.; Mülder, H.; Lankelma, J.; Pinedo, H.; Scheper, R.; Baas, F.; Broxterman, H. The Human Multidrug Resistance-Associated Protein MRP Is a Plasma Membrane Drug-Efflux Pump. Proc. Natl. Acad. Sci. USA 1994, 91, 8822–8826. [Google Scholar] [CrossRef] [Green Version]
- Cole, S.P.; Sparks, K.E.; Fraser, K.; Loe, D.W.; Grant, C.E.; Wilson, G.M.; Deeley, R.G. Pharmacological Characterization of Multidrug Resistant MRP-Transfected Human Tumor Cells. Cancer Res. 1994, 54, 5902–5910. [Google Scholar]
- Loe, D.W.; Deeley, R.; Cole, S. Biology of the Multidrug Resistance-Associated Protein, Mrp. Eur. J. Cancer 1996, 32, 945–957. [Google Scholar] [CrossRef]
- Gao, M.; Yamazaki, M.; Loe, D.W.; Westlake, C.J.; Grant, C.E.; Cole, S.P.; Deeley, R.G. Multidrug Resistance Protein Identification of Regions Required for Active Transport of Leukotriene C4. J. Biol. Chem. 1998, 273, 10733–10740. [Google Scholar] [CrossRef] [Green Version]
- Ohmichi, M.; Hayakawa, J.; Tasaka, K.; Kurachi, H.; Murata, Y. Mechanisms of Platinum Drug Resistance. Trends Pharmacol. Sci. 2005, 26, 113–116. [Google Scholar] [CrossRef]
- Wernyj, R.P.; Morin, P.J. Molecular Mechanisms of Platinum Resistance: Still Searching for the Achilles’Heel. Drug Resist. Updates 2004, 7, 227–232. [Google Scholar] [CrossRef]
- Berretz, G.; Arning, L.; Gerding, W.M.; Friedrich, P.; Fraenz, C.; Schlüter, C.; Epplen, J.T.; Güntürkün, O.; Beste, C.; Genç, E. Structural Asymmetry in the Frontal and Temporal Lobes Is Associated with PCSK6 VNTR Polymorphism. Mol. Neurobiol. 2019, 56, 7765–7773. [Google Scholar] [CrossRef]
- Smedler, E.; Abé, C.; Pålsson, E.; Ingvar, M.; Landén, M. CACNA1C Polymorphism and Brain Cortical Structure in Bipolar Disorder. J. Psychiatry Neurosci. JPN 2019, 45, 190029. [Google Scholar] [PubMed]
- Ross, J.; Hickling, G.; Morgan, D.; Eason, C. The Role of Non-Toxic Prefeed and Postfeed in the Development and Maintenance of 1080 Bait Shyness in Captive Brushtail Possums. Wildl. Res. 2000, 27, 69–74. [Google Scholar] [CrossRef]
- Twigg, L.E. Occurrence of Fluoroacetate in Australian Plants and Tolerance to 1080 in Indigenous Australian Animals. In Proceedings of the Science Workshop, Christchurch, New Zealand; 1994; pp. 97–115. [Google Scholar]
- Kandel, A.; Chenoweth, M.B. Tolerance to Fluoroacetate and Fluorobutyrate in Rats. J. Pharmacol. Exp. Ther. 1952, 104, 248–252. [Google Scholar] [PubMed]
- Guillemaud, T. The Skill and Style to Model the Evolution of Resistance to Pesticides and Drugs. Evol. Appl. 2010, 3, 375–390. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembly Name | Liver Cells | Brain Cells |
|---|---|---|
| Number of contigs | 58,001 | 64,735 |
| Number of contigs ≥ 1000 bp | 13,361 | 12,385 |
| Number of contigs ≥ 5000 bp | 605 | 612 |
| Number of contigs ≥ 10,000 bp | 19 | 24 |
| Total length bp | 47,176,165 | 47,783,051 |
| Total length ≥ 1000 bp | 29,238,918 | 27,088,346 |
| Total length ≥ 5000 bp | 3,994,586 | 4,080,570 |
| Total length ≥ 10,000 bp | 245,362 | 318,579 |
| Largest contig | 18,159 | 21,422 |
| GC % | 42.57 | 42.68 |
| N50 | 1447 | 1247 |
| N75 | 607 | 508 |
| L50 | 8660 | 9524 |
| L75 | 21,240 | 24,749 |
| Metabolic Pathway | Number |
|---|---|
| Global and overview maps | 591 |
| Carbohydrate metabolism | 134 |
| Energy metabolism | 65 |
| Lipid metabolism | 118 |
| Nucleotide metabolism | 12 |
| Amino acid metabolism | 152 |
| Metabolism of other amino acids | 25 |
| Glycan biosynthesis and metabolism | 17 |
| Metabolism of cofactors and vitamins | 54 |
| Metabolism of terpenoids and polyketides | 10 |
| Biosynthesis of other secondary metabolites | 12 |
| Xenobiotics biodegradation and metabolism | 60 |
| Transcription | 8 |
| Translation | 98 |
| Folding, sorting, and degradation | 70 |
| Replication and repair | 3 |
| Membrane transport | 5 |
| Signal transduction | 228 |
| Signalling molecules and interaction | 35 |
| Transport and catabolism | 116 |
| Cell growth and death | 60 |
| Cellular community—eukaryotes | 54 |
| Cellular community—prokaryotes | 2 |
| Cell motility | 23 |
| Immune system | 181 |
| Endocrine system | 185 |
| Circulatory system | 24 |
| Digestive system | 67 |
| Excretory system | 18 |
| Nervous system | 81 |
| Sensory system | 14 |
| Development and regeneration | 21 |
| Aging | 23 |
| Environmental adaptation | 49 |
| Metabolic Pathway | Number |
|---|---|
| Global and overview maps | 254 |
| Carbohydrate metabolism | 62 |
| Energy metabolism | 59 |
| Lipid metabolism | 30 |
| Nucleotide metabolism | 11 |
| Amino acid metabolism | 51 |
| Metabolism of other amino acids | 6 |
| Glycan biosynthesis and metabolism | 10 |
| Metabolism of cofactors and vitamins | 9 |
| Metabolism of terpenoids and polyketides | 5 |
| Biosynthesis of other secondary metabolites | 6 |
| Xenobiotics biodegradation and metabolism | 10 |
| Transcription | 14 |
| Translation | 104 |
| Folding, sorting and degradation | 61 |
| Replication and repair | 6 |
| Membrane transport | 1 |
| Signal transduction | 442 |
| Signaling molecules and interaction | 39 |
| Transport and catabolism | 128 |
| Cell growth and death | 100 |
| Cellular community - eukaryotes | 79 |
| Cellular community - prokaryotes | 2 |
| Cell motility | 32 |
| Immune system | 184 |
| Endocrine system | 256 |
| Circulatory system | 46 |
| Digestive system | 66 |
| Excretory system | 41 |
| Nervous system | 196 |
| Sensory system | 29 |
| Development and regeneration | 44 |
| Aging | 26 |
| Environmental adaptation | 68 |
| Gene Name | Description |
|---|---|
| frmA, ADH5, adhC | S-(hydroxymethyl)glutathione dehydrogenase/alcohol dehydrogenase |
| AOX | aldehyde oxidase |
| DPYD | dihydropyrimidine dehydrogenase (NADP+) |
| MAO, aofH | monoamine oxidase |
| FMO | dimethylaniline monooxygenase (N-oxide forming) |
| UGT | glucuronosyltransferase |
| GST, gst | glutathione S-transferase |
| ndk, NME | nucleoside-diphosphate kinase |
| CES1 | carboxylesterase 1 |
| DPYS, dht, hydA | dihydropyrimidinase |
| dut, DUT | dUTP pyrophosphatase |
| PIK3R1_2_3 | phosphoinositide-3-kinase regulatory subunit α/β/delta |
| CES2 | carboxylesterase 2 |
| CYP1A2 | cytochrome P450 family 1 subfamily A polypeptide 2 |
| CYP2E1 | cytochrome P450 family 2 subfamily E polypeptide 1 |
| GSTK1 | glutathione S-transferase kappa 1 |
| ADH1_7 | alcohol dehydrogenase 1/7 |
| CYP3A4 | cytochrome P450 family 3 subfamily A polypeptide 4 |
| CYP3A5 | cytochrome P450 family 3 subfamily A polypeptide 5 |
| CYP2B6 | cytochrome P450 family 2 subfamily B polypeptide 6 |
| CYP2C8 | cytochrome P450 family 2 subfamily C polypeptide 8 |
| CYP2C19 | cytochrome P450 family 2 subfamily C polypeptide 19 |
| Gene Name | Description |
|---|---|
| CBR1 | carbonyl reductase 1 |
| hprT, hpt, HPRT1 | hypoxanthine phosphoribosyltransferase |
| GST, gst | glutathione S-transferase |
| ndk, NME | nucleoside-diphosphate kinase |
| dut, DUT | dUTP pyrophosphatase |
| PIK3R1_2_3 | phosphoinositide-3-kinase regulatory subunit α/β/delta |
| TOP2 | DNA topoisomerase II |
| ERK, MAPK1_3 | mitogen-activated protein kinase 1/3 |
| AKT RAC | serine/threonine-protein kinase |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emami-Khoyi, A.; Parbhu, S.P.; Ross, J.G.; Murphy, E.C.; Bothwell, J.; Monsanto, D.M.; Vuuren, B.J.v.; Teske, P.R.; Paterson, A.M. De Novo Transcriptome Assembly and Annotation of Liver and Brain Tissues of Common Brushtail Possums (Trichosurus vulpecula) in New Zealand: Transcriptome Diversity after Decades of Population Control. Genes 2020, 11, 436. https://doi.org/10.3390/genes11040436
Emami-Khoyi A, Parbhu SP, Ross JG, Murphy EC, Bothwell J, Monsanto DM, Vuuren BJv, Teske PR, Paterson AM. De Novo Transcriptome Assembly and Annotation of Liver and Brain Tissues of Common Brushtail Possums (Trichosurus vulpecula) in New Zealand: Transcriptome Diversity after Decades of Population Control. Genes. 2020; 11(4):436. https://doi.org/10.3390/genes11040436
Chicago/Turabian StyleEmami-Khoyi, Arsalan, Shilpa Pradeep Parbhu, James G. Ross, Elaine C. Murphy, Jennifer Bothwell, Daniela M. Monsanto, Bettine Jansen van Vuuren, Peter R. Teske, and Adrian M. Paterson. 2020. "De Novo Transcriptome Assembly and Annotation of Liver and Brain Tissues of Common Brushtail Possums (Trichosurus vulpecula) in New Zealand: Transcriptome Diversity after Decades of Population Control" Genes 11, no. 4: 436. https://doi.org/10.3390/genes11040436