
Pendred Syndrome, or Not Pendred Syndrome? That Is the Question

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples Collection
2.2. DNA Extraction and Quantification
2.3. GJB2 and GJB6 Analyses
2.4. Whole Exome Sequencing (WES)
2.5. CEVA Haplotype Analysis
2.6. Multiplex Ligation Probe Amplification (MLPA)
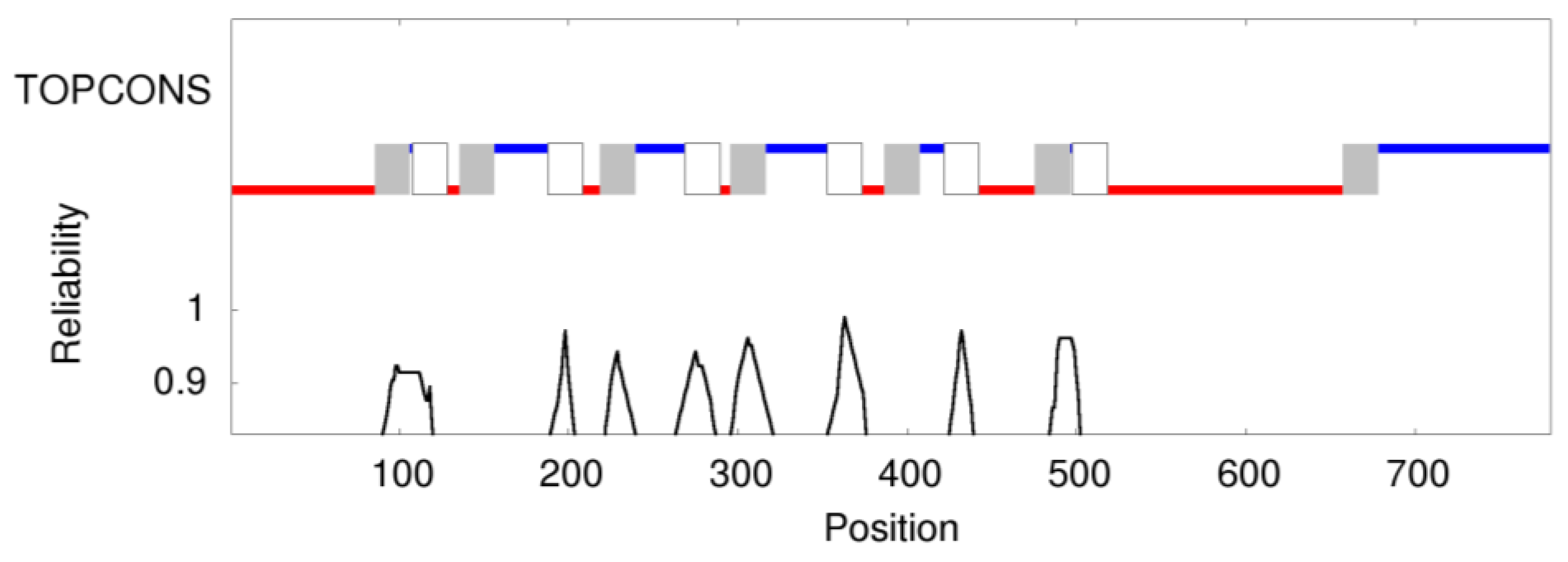
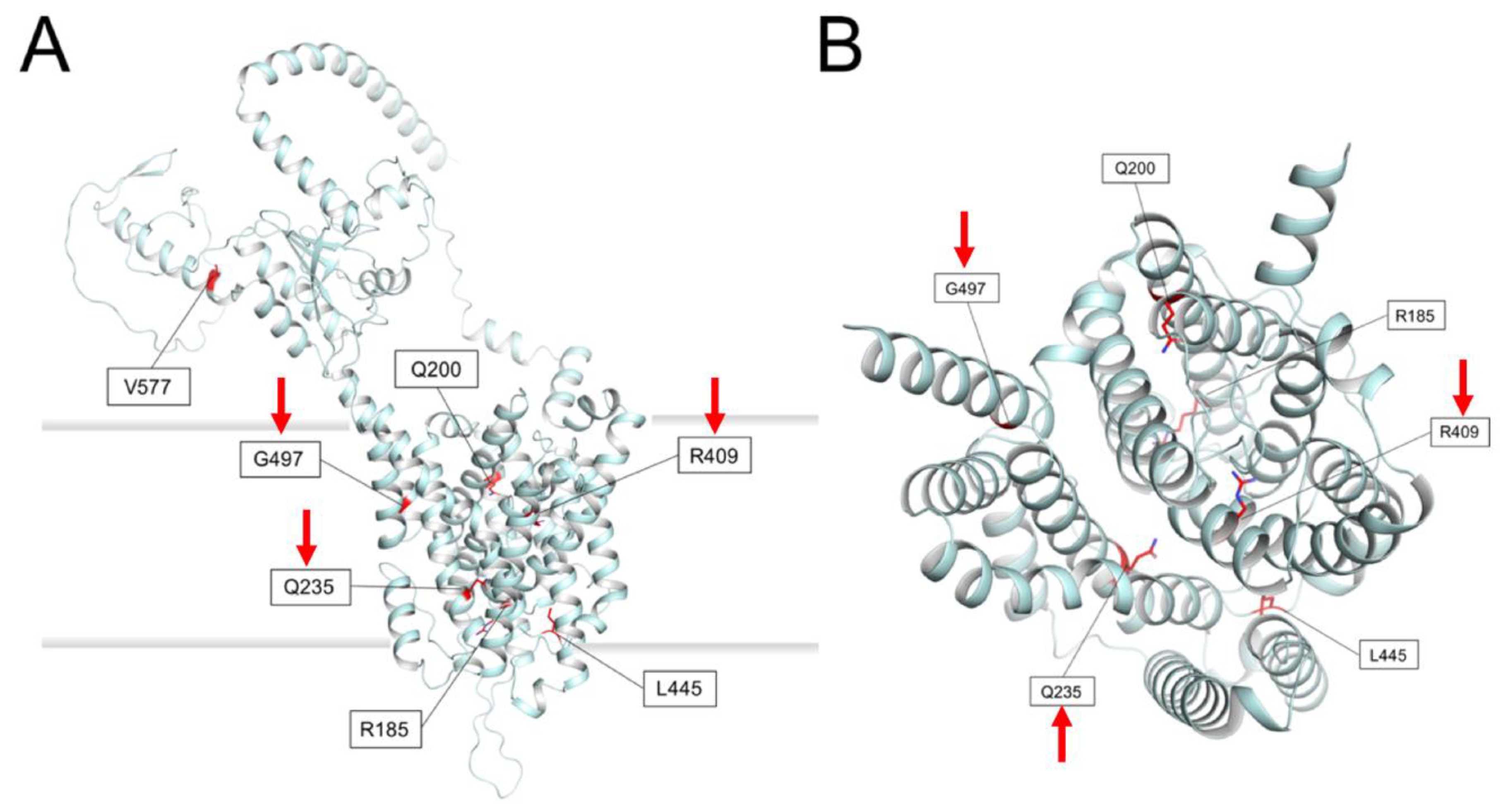
2.7. Prediction of Membrane Topology and 3D Molecular Model of Pendrin
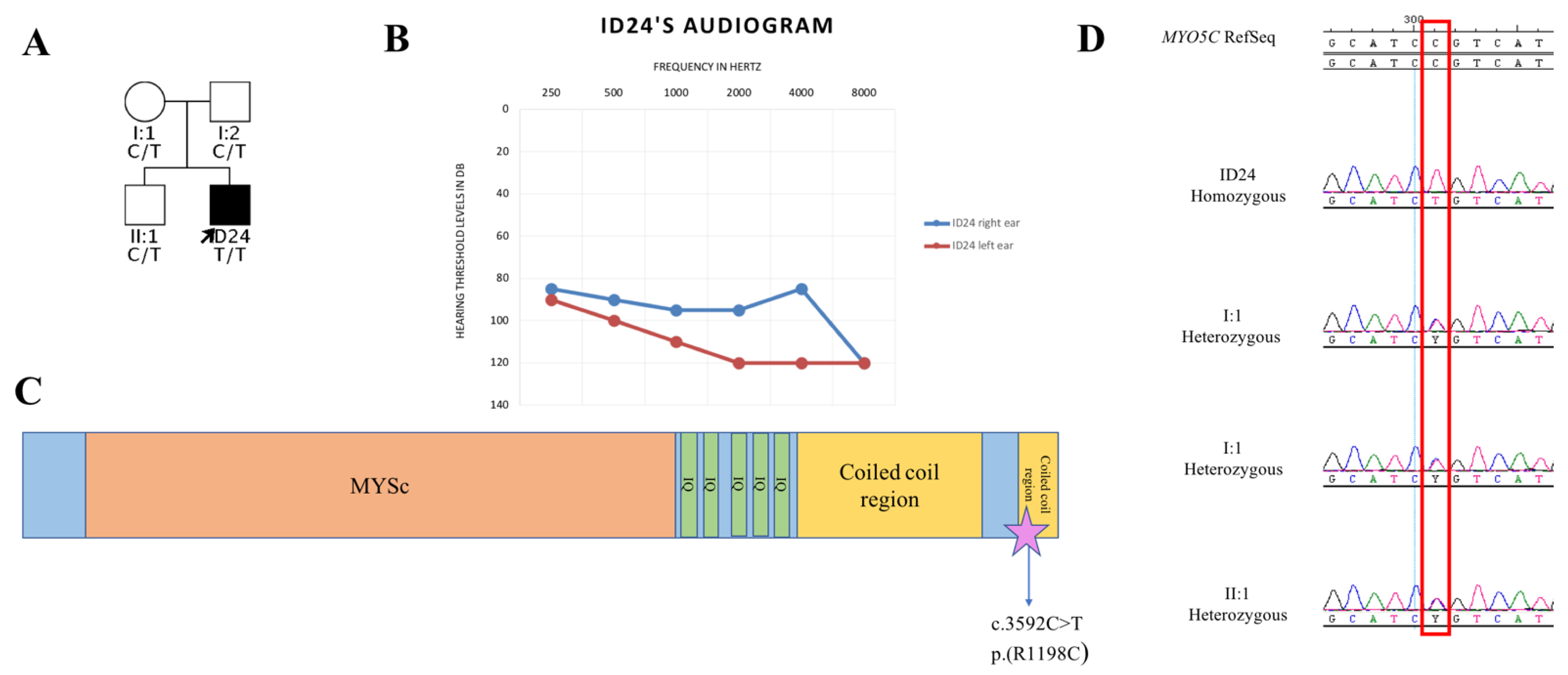
3. Results
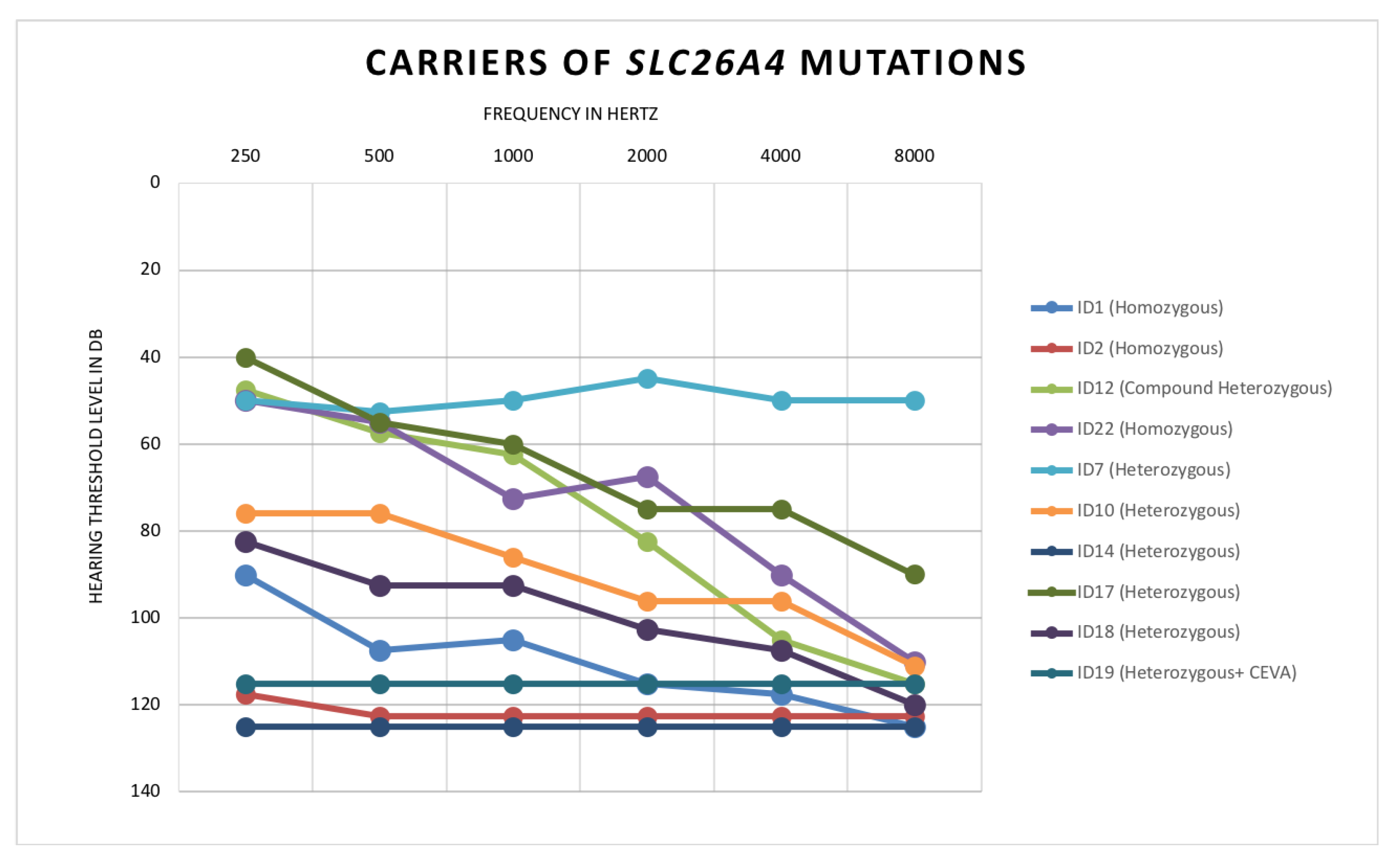
- Patient ID1: Homozygous for c.1225C>T, p.(R409C)
- Patient ID2: Homozygous for c.704A>G, p.(Q235R)
- Patient ID22: Homozygous for c.1489G>A, p.(G497S)
- Patient ID12: Compound heterozygous for c.1001+1G>A and c.1149+3A>G
- ID7: Heterozygous c.1536_1537delAG, p.(R512Sfs*14)
- ID10: Heterozygous c.1489G>A, p.(G497S)
- ID14: Heterozygous c.554G>C, p.(R185T)
- ID17: Heterozygous c.1263+2T>C
- ID18: Heterozygous c.1730T>C, p. (V577A)
- ID19: Heterozygous c.600G>A, p.(Q200Q)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Korver, A.M.; Smith, R.J.; Van Camp, G.; Schleiss, M.R.; Bitner-Glindzicz, M.A.; Lustig, L.R.; Usami, S.I.; Boudewyns, A.N. Congenital hearing loss HHS Public Access. Nat. Rev. Dis. Prim. 2018, 3, 1–37. [Google Scholar] [CrossRef]
- Casazza, G.; Meier, J.D. Evaluation and management of syndromic congenital hearing loss. Curr. Opin. Otolaryngol. Head Neck Surg. 2017, 25, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.B.; Goldman, J. Syndromic Sensorineural Hearing Loss. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK526088/ (accessed on 24 August 2020).
- De Lima, Y.S.; Chiabai, M.; Shen, J.; Córdoba, M.S.; Versiani, B.R.; Benício, R.O.A.; Pogue, R.; Mingroni-Netto, R.C.; Lezirovitz, K.; Pic-Taylor, A.; et al. Syndromic hearing loss molecular diagnosis: Application of massive parallel sequencing. Hear Res. 2018, 370, 181–188. [Google Scholar] [CrossRef]
- Ideura, M.; Nishio, S.Y.; Moteki, H.; Takumi, Y.; Miyagawa, M.; Sato, T.; Kobayashi, Y.; Ohyama, K.; Oda, K.; Matsui, T.; et al. Comprehensive analysis of syndromic hearing loss patients in Japan. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wémeau, J.L.; Kopp, P. Pendred syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Bizhanova, A.; Kopp, P. Genetics and phenomics of Pendred syndrome. Mol. Cell Endocrinol. 2010, 322, 83–90. [Google Scholar] [CrossRef]
- Forli, F.; Lazzerini, F.; Auletta, G.; Bruschini, L.; Berrettini, S. Enlarged vestibular aqueduct and Mondini Malformation: Audiological, clinical, radiologic and genetic features. Eur. Arch. Oto-Rhino-Laryngol. 2021, 278, 2305–2312. [Google Scholar] [CrossRef]
- Soh, L.M.; Druce, M.; Grossman, A.B.; Differ, A.M.; Rajput, L.; Bitner-Glindzicz, M.; Korbonits, M. Evaluation of genotype-phenotype relationships in patients referred for endocrine assessment in suspected Pendred syndrome. Eur. J. Endocrinol. 2015, 172, 217–226. [Google Scholar] [CrossRef]
- Suzuki, H.; Oshima, A.; Tsukamoto, K.; Abe, S.; Kumakawa, K.; Nagai, K.; Satoh, H.; Kanda, Y.; Iwasaki, S.; Usami, S. Clinical characteristics and genotype-phenotype correlation of hearing loss patients with SLC26A4 mutations. Acta Otolaryngol. 2007, 127, 1292–1297. [Google Scholar] [CrossRef]
- Reardon, W.; Coffey, R.; Chowdhury, T.; Grossman, A.; Jan, H.; Britton, K.; Kendall-Taylor, P.; Trembath, R. Prevalence, age of onset, and natural history of thyroid disease in Pendred syndrome. J. Med. Genet. 1999, 36, 595–598. [Google Scholar] [CrossRef]
- Garabet Diramerian, L.; Ejaz, S. Pendred Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK549839/ (accessed on 30 June 2021).
- Smith, R.J.H.; Iwasa, Y.; Schaefer, A.M. Pendred Syndrome/Nonsyndromic Enlarged Vestibular Aqueduct. 28 September 1998. In Adam MP, GeneReviews® [Internet]; Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 1993–2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1467/ (accessed on 18 June 2020).
- Li, M.; Nishio, S.Y.; Naruse, C.; Riddell, M.; Sapski, S.; Katsuno, T.; Hikita, T.; Mizapourshafiyi, F.; Smith, F.M.; Cooper, L.T.; et al. Digenic inheritance of mutations in EPHA2 and SLC26A4 in Pendred syndrome. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Griffith, A.J. Genetic architecture and phenotypic landscape of SLC26A4-related hearing loss. Hum. Genet. 2021, 1–10. [Google Scholar] [CrossRef]
- Dewan, K.; Wippold, F.J., II; Lieu, J.E.C. Enlarged vestibular aqueduct in pediatric SNHL. Otolaryngol. Head Neck Surg. 2009, 140, 552. [Google Scholar] [CrossRef]
- Sarioglu, F.C.; Cakir Cetin, A.; Guleryuz, H.; Guneri, E.A. The Diagnostic Efficacy of MRI in the Evaluation of the Enlarged Vestibular Aqueduct in Children with Hearing Loss. Turki. Arch. Otorhinolaryngol. 2021, 58, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Maravall, F.J.; Gómez-Arnáiz, N.; Gumá, A.; Abós, R.; Soler, J.; Gómez, J.M. Reference values of thyroid volume in a healthy, non-iodine-deficient Spanish population. Horm. Metab. Res. 2004, 36, 645–649. [Google Scholar] [CrossRef]
- Berghout, A.; Wiersinga, W.M.; Smits, N.J.; Touber, J.L. Determinants of thyroid volume as measured by ultrasonography in healthy adults in a non-iodine deficient area. Clin. Endocrinol. 1987, 26, 273–280. [Google Scholar] [CrossRef]
- Del Castillo, I.; Villamar, M.; Moreno-Pelayo, M.A.; del Castillo, F.J.; Alvarez, A.; Tellería, D.; Menéndez, I.; Moreno, F. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N. Engl. J. Med. 2002, 346, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limongelli, I.; Marini, S.; Bellazzi, R. PaPI: Pseudo amino acid composition to score human protein-coding variants. BMC Bioinform. 2015, 16, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, X.; Boerwinkle, E.; Liu, X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014, 42, 13534–13544. [Google Scholar] [CrossRef] [Green Version]
- Tsirigos, K.D.; Peters, C.; Shu, N.; Käll, L.; Elofsson, A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015, 43, W401–W407. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Chi, X.; Jin, X.; Chen, Y.; Lu, X.; Tu, X.; Li, X.; Zhang, Y.; Lei, J.; Huang, J.; Huang, Z.; et al. Structural insights into the gating mechanism of human SLC26A9 mediated by its C-terminal sequence. Cell Discov. 2020, 6, 1–10. [Google Scholar] [CrossRef]
- Bassot, C.; Minervini, G.; Leonardi, E.; Tosatto, S.C.E. Mapping pathogenic mutations suggests an innovative structural model for the pendrin (SLC26A4) transmembrane domain. Biochimie 2017, 132, 109–120. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. Mutationtaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Busi, M.; Rosignoli, M.; Castiglione, A.; Minazzi, F.; Trevisi, P.; Aimoni, C.; Calzolari, F.; Granieri, E.; Martini, A. Cochlear Implant Outcomes and Genetic Mutations in Children with Ear and Brain Anomalies. Biomed. Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coyle, B.; Reardon, W.; Herbrick, J.A.; Tsui, L.C.; Gausden, E.; Lee, J.; Coffey, R.; Grueters, A.; Grossman, A.; Phelps, P.D.; et al. Molecular analysis of the PDS gene in Pendred syndrome (sensorineural hearing loss and goitre). Hum. Mol. Genet. 1998, 7, 1105–1112. [Google Scholar] [CrossRef] [Green Version]
- Wasano, K.; Takahashi, S.; Rosenberg, S.K.; Kojima, T.; Mutai, H.; Matsunaga, T.; Ogawa, K.; Homma, K. Systematic quantification of the anion transport function of pendrin (SLC26A4) and its disease-associated variants. Hum. Mutat. 2020, 41, 316–331. [Google Scholar] [CrossRef]
- Downie, L.; Halliday, J.; Burt, R.; Lunke, S.; Lynch, E.; Martyn, M.; Poulakis, Z.; Gaff, C.; Sung, V.; Wake, M.; et al. Exome sequencing in infants with congenital hearing impairment: A population-based cohort study. Eur. J. Hum. Genet. 2020, 28, 587–596. [Google Scholar] [CrossRef]
- Lee, B.; Kim, Y.R.; Kim, S.J.; Goh, S.H.; Kim, J.H.; Oh, S.K.; Baek, J.I.; Kim, U.K.; Lee, K.Y. Modified U1 snRNA and antisense oligonucleotides rescue splice mutations in SLC26A4 that cause hereditary hearing loss. Hum. Mutat. 2019, 40, 1172–1180. [Google Scholar] [CrossRef] [PubMed]
- Lenarduzzi, S.; Morgan, A.; Faletra, F.; Cappellani, S.; Morgutti, M.; Mezzavilla, M.; Peruzzi, A.; Ghiselli, S.; Ambrosetti, U.; Graziano, C.; et al. Next generation sequencing study in a cohort of Italian patients with syndromic hearing loss. Hear. Res. 2019, 381, 107769. [Google Scholar] [CrossRef]
- Roesch, S.; Bernardinelli, E.; Nofziger, C.; Tóth, M.; Patsch, W.; Rasp, G.; Paulmichl, M.; Dossena, S. Functional testing of SLC26A4 variants—clinical and molecular analysis of a cohort with enlarged vestibular aqueduct from Austria. Int. J. Mol. Sci. 2018, 19, 209. [Google Scholar] [CrossRef] [Green Version]
- Hosoya, M.; Fujioka, M.; Sone, T.; Okamoto, S.; Akamatsu, W.; Ukai, H.; Ueda, H.R.; Ogawa, K.; Matsunaga, T.; Okano, H. Cochlear Cell Modeling Using Disease-Specific iPSCs Unveils a Degenerative Phenotype and Suggests Treatments for Congenital Progressive Hearing Loss. Cell Rep. 2017, 18, 68–81. [Google Scholar] [CrossRef]
- Charfeddine, I.; Mnejja, M.; Hammami, B.; Chakroun, A.; Masmoudi, S.; Ayadi, H.; Ghorbel, A. Pendred syndrome in Tunisia. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2010, 127, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Bigozzi, M.; Melchionda, S.; Casano, R.; Palladino, T.; Gitti, G. Pendred syndrome: Study of three families. Acta Otorhinolaryngol. Ital. 2005, 25, 233–239. [Google Scholar]
- Huang, C.J.; Lei, T.H.; Chang, W.L.; Tu, T.Y.; Shiao, A.S.; Chiu, C.Y.; Jap, T.S. A Novel mutation in the SLC26A4 gene in a Chinese family with Pendred syndrome. Int. J. Pediatr. Otorhinolaryngol. 2013, 77, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Aimoni, C.; Ciorba, A.; Cerritelli, L.; Ceruti, S.; Skarżyński, P.H.; Hatzopoulos, S. Enlarged vestibular aqueduct: Audiological and genetical features in children and adolescents. Int. J. Pediatric Otorhinolaryngol. 2017, 101, 254–258. [Google Scholar] [CrossRef] [PubMed]
- López-Bigas, N.; Rabionet, R.; de Cid, R.; Govea, N.; Gasparini, P.; Zelante, L.; Arbonés, M.L.; Estivill, X. Splice-site mutation in the PDS gene may result in intrafamilial variability for deafness in pendred syndrome. Hum. Mutat. 1999, 14, 520–526. [Google Scholar] [CrossRef]
- Rodriguez, O.C.; Cheney, R.E. Human myosin-Vc is a novel class V myosin expressed in epithelial cells. J. Cell Sci. 2002, 115, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, D.T.; Weigert, R.; Grode, K.D.; Donaldson, J.G.; Cheney, R.E. Myosin Vc is a molecular motor that functions in secretory granule trafficking. Mol. Biol. Cell. 2009, 20, 4471–4488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, T.B.; Belyantseva, I.A.; Frolenkov, G.I. Myosins and Hearing. Adv. Exp. Med. Biol. 2020, 1239, 317–330. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age (Years Old) | EVA | Severity of the Malformation | ESE | Cochlear Abnormalities (IP-II) | Semicircular Canal Vestibular Abnormalities | Average Hearing Threshold | Classification |
|---|---|---|---|---|---|---|---|---|
| ID1 | 29 | Bilateral | Moderate | Bilateral | Bilateral | Bilateral | Profound | M2 |
| ID2 | 2 | Bilateral | Moderate | Bilateral | Bilateral | Bilateral | Profound | M2 |
| ID3 | 11 | Bilateral | Moderate | Bilateral | Bilateral | Bilateral | Severe | NA |
| ID4 | 3 | Bilateral | Mild | Bilateral | Bilateral | Bilateral | Mild | NA |
| ID5 | 14 | Bilateral | Moderate | Bilateral | Bilateral | Bilateral | Moderate | NA |
| ID6 | 13 | Bilateral | Moderate | Bilateral | NO | Bilateral | Moderate | NA |
| ID7 | 12 | Bilateral | Moderate | Bilateral | Bilateral | Bilateral | Moderate | M1 |
| ID8 | 13 | Bilateral | Mild | NO | NO | Bilateral | Moderate | NA |
| ID9 | 21 | Left | Mild | NO | IP-II Left | Bilateral | Moderate | NA |
| ID10 | 10 | Left | Mild | Left | IP-II Left | NA | Profound | M1 |
| ID11 | 30 | Bilateral | Moderate | Bilateral | Bilateral | Bilateral | Severe | NA |
| ID12 | 12 | Bilateral | Mild | NA | NA | NA | Severe | M2 |
| ID13 | 7 | Bilateral | Mild | Bilateral | Bilateral | Bilateral | Severe | NA |
| ID14 | 6 | Bilateral | Mild | NO | Bilateral | Bilateral | Profound | M1 |
| ID15 | 13 | Bilateral | Moderate | Bilateral | Bilateral | NO | Moderate | NA |
| ID16 | 6 | Bilateral | Moderate | Bilateral | Bilateral | NO | Moderate | NA |
| ID17 | 15 | Bilateral | Moderate | Bilateral | Bilateral | Bilateral | Moderate | M1 |
| ID18 | 7 | Bilateral | NA | NA | NA | NA | Profound | M1 |
| ID19 | 16 | Bilateral | Moderate | NA | Bilateral | Bilateral | Profound | M2 |
| ID20 | 10 | Bilateral | Mild | NA | Bilateral | Bilateral | Moderate | NA |
| ID21 | 23 | Bilateral | Mild | Bilateral | Bilateral | Bilateral | Moderate | NA |
| ID22 | 29 | Bilateral | NA | Bilateral | Bilateral | Bilateral | Severe | M2 |
| ID23 | 1 | Bilateral | Mild | Left | NA | NA | Severe | NA |
| ID24 | 13 | Bilateral | Moderate | NA | Bilateral | Bilateral | Profound | NA |
| Patient | SLC26A4 cDNA Change | Classification | Protein Change | dbSNP | GnomAD | PolyPhen [22] | SIFT [23] | Mutation Taster [31] | Reference |
|---|---|---|---|---|---|---|---|---|---|
| ID1 | c.1225C>T (Hom) | M2 | p.(R409C) | rs147952620 | 0.001594% | 1 (d) | 0 (d) | d. c. | [30] |
| ID2 | c.704A>T (Hom) | M2 | p.(Q235R) | rs752485540 | 0.0007357% | 1 (d) | 0 (d) | d. c. | [32] |
| ID7 | c.1536_1537delAG (Het) | M1 | p.(R512Sfs*14) | rs1435734312 | NA | NA | NA | d. c. | [33] |
| ID10 | c.1489G>A (Het) | M1 | p.(G497S) | rs111033308 | 0.002475% | 1 (d) | 0 (d) | d. c. | [34] |
| ID12 | c.1001+1G>A (Het) | M2 | NA | rs80338849 | 0.02059% | NA | NA | d.c. | [35] |
| c.1149+3A>G (Het) | NA | rs111033314 | 0.002389% | NA | NA | d.c. | [36] | ||
| ID14 | c.554G>C (Het) | M1 | p.(R185T) | rs542620119 | 0.008485% | 0.98 (d) | 0.04 (d) | d. c. | [30] |
| ID17 | c.1263+2T>C (Het) | M1 | NA | NA | NA | NA | NA | d.c. | [37] |
| ID18 | c.1730T>C (Het) | M1 | p. (V577A) | rs56017519 | 0.0003982% | 0.999 (d) | 0.1 (t) | d. c. | [38] |
| ID19 | c.600G>A (Het) | M2 (CEVA) | p.(Q200Q) | NA | NA | NA | NA | d. c. | NA |
| ID22 | c.1489G>A (Hom) | M2 | p.(G497S) | rs111033308 | 0.002475% | 1 (d) | 0 (d) | d. c. | [34] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tesolin, P.; Fiorino, S.; Lenarduzzi, S.; Rubinato, E.; Cattaruzzi, E.; Ammar, L.; Castro, V.; Orzan, E.; Granata, C.; Dell’Orco, D.; et al. Pendred Syndrome, or Not Pendred Syndrome? That Is the Question. Genes 2021, 12, 1569. https://doi.org/10.3390/genes12101569
Tesolin P, Fiorino S, Lenarduzzi S, Rubinato E, Cattaruzzi E, Ammar L, Castro V, Orzan E, Granata C, Dell’Orco D, et al. Pendred Syndrome, or Not Pendred Syndrome? That Is the Question. Genes. 2021; 12(10):1569. https://doi.org/10.3390/genes12101569
Chicago/Turabian StyleTesolin, Paola, Sofia Fiorino, Stefania Lenarduzzi, Elisa Rubinato, Elisabetta Cattaruzzi, Lydie Ammar, Veronica Castro, Eva Orzan, Claudio Granata, Daniele Dell’Orco, and et al. 2021. "Pendred Syndrome, or Not Pendred Syndrome? That Is the Question" Genes 12, no. 10: 1569. https://doi.org/10.3390/genes12101569
APA StyleTesolin, P., Fiorino, S., Lenarduzzi, S., Rubinato, E., Cattaruzzi, E., Ammar, L., Castro, V., Orzan, E., Granata, C., Dell’Orco, D., Morgan, A., & Girotto, G. (2021). Pendred Syndrome, or Not Pendred Syndrome? That Is the Question. Genes, 12(10), 1569. https://doi.org/10.3390/genes12101569