
Angiotensin–Converting Enzyme (ACE) 1 Gene Polymorphism and Phenotypic Expression of COVID-19 Symptoms


Abstract
:1. Introduction
2. Physiological and Pathological Roles of the RAAS
3. Roles of ACEs and Ang II in the Immune System
4. Possible Involvement of ACE1 I/D Polymorphism in the Aggravation of COVID-19 Symptoms
4.1. Literature and Database Searches (Epidemiological Studies)
4.2. Studies with Patient Samples (Clinical Studies)
4.3. Possible Involvement of Polymorphic Alu Elements and Their Functional Aspects
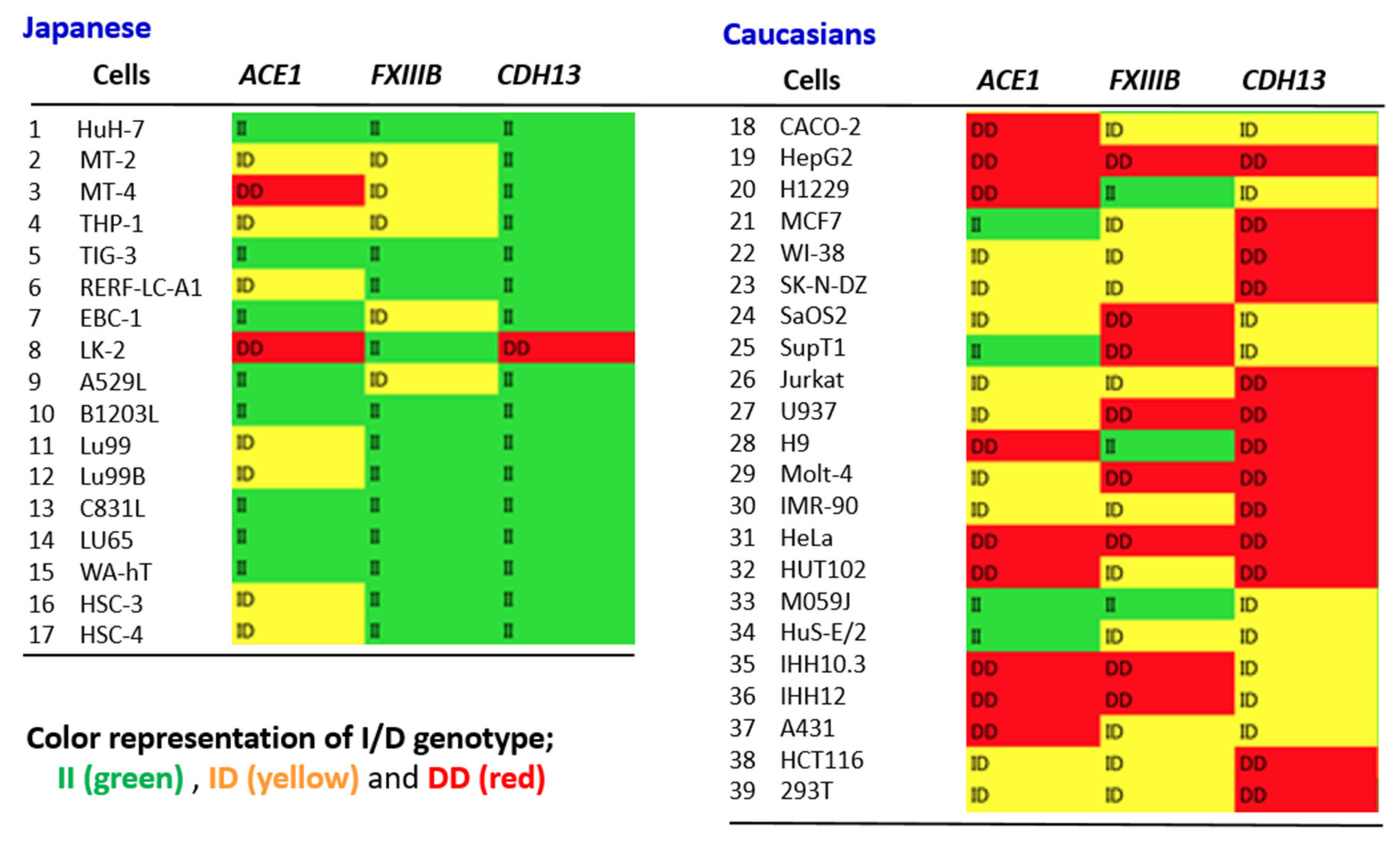
4.4. Possible Interplay between ACE1 and Other Alu Variants
4.5. ACE1 I/D Genotype Likely to Match Other Alu Gene Polymorphisms at the Individual Level
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAT | alfa-1 anti-trypsin |
| ABC transporters | ATP-binding cassette transporters |
| ACE | angiotensin-converting enzyme |
| ADAM17 | ADAM metallopeptidase domain 17 |
| Ang | angiotensin |
| ARDS | acute respiratory distress syndrome |
| HLA | human leukocyte antigen |
| IL | interleukin |
| LGI | low-grade inflammation |
| Nsps | non-structural proteins |
| NF-kB | nuclear factor kappa B |
| Orf | open reading frame |
| PLAT | plasminogen activator |
| PAI-1 | plasminogen activator inhibitor-1 |
| PKC | Protein kinase C |
| RAAS | renin–angiotensin–aldosterone system |
| ROS | reactive oxygen species |
| SARS | severe acute respiratory syndrome |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| STAT | signal transducer and activator of transcription |
| TNF-α | tumor necrosis factor-α |
| tPA | tissue-type plasminogen activator |
References
- Nguyen, H.T.; Zhang, S.; Wang, Q.; Anang, S.; Wang, J.; Ding, H.; Kappes, J.C.; Sodroski, J. Spike glycoprotein and host cell determinants of SARS-CoV-2 entry and cytopathic effects. J. Virol. 2020, 95, e02304–e02320. [Google Scholar]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Biomed. 2020, 91, 157–160. [Google Scholar] [PubMed]
- WHO. WHO Coronavirus (COVID-19) Dashboard. 2021. Available online: https://covid19.who.int/ (accessed on 6 August 2021).
- Yamamoto, N.; Ariumi, Y.; Nishida, N.; Yamamoto, R.; Bauer, G.; Gojobori, T.; Shimotohno, K.; Mizokami, M. SARS-CoV-2 infections and COVID-19 mortalities strongly correlate with ACE1 I/D genotype. Gene 2020, 758, 144944. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.J.; Jiang, S. The spike protein of SARS-CoV—A target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef]
- Ko, C.-J.; Huang, C.-C.; Lin, H.-Y.; Juan, C.-P.; Lan, S.-W.; Shyu, H.-Y.; Wu, S.-R.; Hsiao, P.-W.; Huang, H.-P.; Shun, C.-T.; et al. Androgen-Induced TMPRSS2 Activates Matriptase and Promotes Extracellular Matrix Degradation, Prostate Cancer Cell Invasion, Tumor Growth, and Metastasis. Cancer Res. 2015, 75, 2949–2960. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Wang, Y.; Ou, L.; Li, J.; Zheng, K.; Zhan, H.; Gu, J.; Zhou, G.; Xie, S.; Zhang, J.; et al. Downregulation of ACE2 expression by SARS-CoV-2 worsens the prognosis of KIRC and KIRP patients via metabolism and immunoregulation. Int. J. Biol. Sci. 2021, 17, 1925–1939. [Google Scholar] [CrossRef]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef]
- Bansal, M. Cardiovascular disease and COVID-19. Diabetes Metab. Syndr. 2020, 14, 247–250. [Google Scholar] [CrossRef]
- Nossent, E.J.; Schuurman, A.R.; Reijnders, T.D.; Saris, A.; Jongerius, I.; Blok, S.G.; de Vries, H.; Duitman, J.; Noordegraaf, A.V.; Meijboom, L.J.; et al. Pulmonary Procoagulant and Innate Immune Responses in Critically Ill COVID-19 Patients. Front. Immunol. 2021, 12, 664209. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Dass, J.; Mahapatra, M. Hemostatic Abnormalities in COVID-19: An Update. Indian J. Hematol. Blood Transfus. 2020, 36, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.H.; Gallus, A.; Jindal, R.; Wang, C.; Wu, C.C. Incidence of Venous Thromboembolism in Asian Populations: A Systematic Review. Thromb. Haemost. 2017, 117, 2243–2260. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Murakami, M. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release Syndrome. Immunity 2020, 52, 731–733. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Wang, R.; Feng, Z.; Zhang, J.; Yang, H.; Tan, Y.; Wang, H.; Wang, C.; Liu, L.; et al. Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 infection. Nat. Commun. 2021, 12, 2506. [Google Scholar] [CrossRef]
- Gao, Y.-D.; Ding, M.; Dong, X.; Zhang, J.-J.; Azkur, A.K.; Azkur, D.; Gan, H.; Sun, Y.-L.; Fu, W.; Li, W.; et al. Risk factors for severe and critically ill COVID-19 patients: A review. Allergy 2021, 76, 428–455. [Google Scholar] [CrossRef]
- Yamamoto, N.; Yamamoto, R.; Ariumi, Y.; Mizokami, M.; Shimotohno, K.; Yoshikura, H. Does Genetic Predisposition Contribute to the Exacerbation of COVID-19 Symptoms in Individuals with Comorbidities and Explain the Huge Mortality Disparity between the East and the West? Int. J. Mol. Sci. 2021, 22, 5000. [Google Scholar] [CrossRef]
- Lund, M.A.V.; Thostrup, A.H.; Frithioff-Bojsoe, C.; Lausten-Thomsen, U.; Hedley, P.L.; Pedersen, O.; Christiansen, M.; Hansen, T.; Holm, J.-C. Low-grade inflammation independently associates with cardiometabolic risk in children with overweight/obesity. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1544–1553. [Google Scholar] [CrossRef]
- Russo, L.; Lumeng, C.N. Properties and functions of adipose tissue macrophages in obesity. Immunology 2018, 155, 407–417. [Google Scholar] [CrossRef]
- Scully, E.P.; Haverfield, J.; Ursin, R.L.; Tannenbaum, C.; Klein, S.L. Considering how biological sex impacts immune responses and COVID-19 outcomes. Nat. Rev. Immunol. 2020, 20, 442–447. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Fajnzylber, J.; Regan, J.; Coxen, K.; Corry, H.; Wong, C.; Rosenthal, A.; Worrall, D.; Giguel, F.; Piechocka-Trocha, A.; Atyeo, C.; et al. SARS-CoV-2 viral load is associated with increased disease severity and mortality. Nat. Commun. 2020, 11, 5493. [Google Scholar] [CrossRef]
- Te Riet, L.; van Esch, J.H.; Roks, A.J.; van den Meiracker, A.H.; Danser, A.H. Hypertension: Renin-angiotensin-aldosterone system alterations. Circ. Res. 2015, 116, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Senchenkova, E.Y.; Russell, J.; Vital, S.A.; Yildirim, A.; Orr, A.W.; Granger, D.N.; Gavins, F.N.E. A critical role for both CD40 and VLA5 in angiotensin II-mediated thrombosis and inflammation. FASEB J. 2018, 32, 3448–3456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Li, S.; Niu, S. ACE2 and COVID-19 and the resulting ARDS. Postgrad. Med. J. 2020, 96, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Wolin, M.S.; Csiszar, A. Mechanosensitive production of reactive oxygen species in endothelial and smooth muscle cells: Role in microvascular remodeling? Antioxid. Redox Signal. 2006, 8, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.J.; Vapaatalo, H.; Mervaala, E. Angiotensin II and vascular inflammation. Med. Sci. Monit. 2005, 11, RA194–RA205. [Google Scholar] [PubMed]
- Luft, F.C.; Dechend, R.; Muller, D.N. Immune mechanisms in angiotensin II-induced target-organ damage. Ann. Med. 2012, 44, S49–S54. [Google Scholar] [CrossRef] [PubMed]
- Larsson, P.T.; Schwieler, J.H.; Wallen, N.H. Platelet activation during angiotensin II infusion in healthy volunteers. Blood Coagul. Fibrinolysis 2000, 11, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Mian, M.O.; Barhoumi, T.; Briet, M.; Paradis, P.; Schiffrin, E.L. Deficiency of T-regulatory cells exaggerates angiotensin II-induced microvascular injury by enhancing immune responses. J. Hypertens. 2016, 34, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.P.; Vinh, A.; Tuck, K.; Sakkal, S.; Krishnan, S.M.; Chan, C.T.; Lieu, M.; Samuel, C.S.; Diep, H.; Kemp-Harper, B.K.; et al. M2 macrophage accumulation in the aortic wall during angiotensin II infusion in mice is associated with fibrosis, elastin loss, and elevated blood pressure. Am. J. Physiol. Circ. Physiol. 2015, 309, H906–H917. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.M.; França-Falcão, M.S.; Calzerra, N.T.M.; Luz, M.S.; Gadelha, D.D.A.; Balarini, C.M.; Queiroz, T.M. Role of Renin-Angiotensin System Components in Atherosclerosis: Focus on Ang-II, ACE2, and Ang-1–7. Front. Physiol. 2020, 11, 1067. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Zhang, Y.-H.; Dong, X.-F.; Hao, Q.-Q.; Zhou, X.-M.; Yu, Q.-T.; Li, S.-Y.; Chen, X.; Tengbeh, A.F.; Dong, B.; et al. ACE2 and Ang-(1–7) protect endothelial cell function and prevent early atherosclerosis by inhibiting inflammatory response. Inflamm. Res. 2015, 64, 253–260. [Google Scholar] [CrossRef]
- Brown, N.J.; Vaughan, D.E. Prothrombotic effects of angiotensin. Adv. Intern. Med. 2000, 45, 419–429. [Google Scholar] [PubMed]
- Fogari, R.; Zoppi, A.; Mugellini, A.; Maffioli, P.; Lazzari, P.; Derosa, G. Role of angiotensin II in plasma PAI-1 changes induced by imidapril or candesartan in hypertensive patients with metabolic syndrome. Hypertens. Res. 2011, 34, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Gando, S.; Wada, T. Thromboplasminflammation in COVID-19 Coagulopathy: Three Viewpoints for Diagnostic and Therapeutic Strategies. Front. Immunol. 2021, 12, 649122. [Google Scholar] [CrossRef]
- Marshall, R.P.; Webb, S.; Bellingan, G.J.; Montgomery, H.E.; Chaudhari, B.; McAnulty, R.J.; Humphries, S.E.; Hill, M.R.; Laurent, G.J. Angiotensin Converting Enzyme Insertion/Deletion Polymorphism Is Associated with Susceptibility and Outcome in Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2002, 166, 646–650. [Google Scholar] [CrossRef]
- Itoyama, S.; Keicho, N.; Quy, T.; Phi, N.C.; Long, H.T.; Ha, L.D.; Van Ban, V.; Ohashi, J.; Hijikata, M.; Matsushita, I.; et al. ACE1 polymorphism and progression of SARS. Biochem. Biophys. Res. Commun. 2004, 323, 1124–1129. [Google Scholar] [CrossRef]
- Pabalan, N.; Tharabenjasin, P.; Suntornsaratoon, P.; Jarjanazi, H.; Muanprasat, C. Ethnic and age-specific acute lung injury/acute respiratory distress syndrome risk associated with angiotensin-converting enzyme insertion/deletion polymorphisms, implications for COVID-19: A meta-analysis. Infect. Genet. Evol. 2021, 88, 104682. [Google Scholar] [CrossRef] [PubMed]
- Pati, A.; Mahto, H.; Padhi, S.; Panda, A.K. ACE deletion allele is associated with susceptibility to SARS-CoV-2 infection and mortality rate: An epidemiological study in the Asian population. Clin. Chim. Acta 2020, 510, 455–458. [Google Scholar] [CrossRef]
- Aung, A.K.; Aitken, T.; Teh, B.M.; Yu, C.; Ofori-Asenso, R.; Chin, K.L.; Liew, D. Angiotensin converting enzyme genotypes and mortality from COVID-19: An ecological study. J. Infect. 2020, 81, 961–965. [Google Scholar] [CrossRef]
- Bellone, M.; Calvisi, S.L. ACE polymorphisms and COVID-19-related mortality in Europe. J. Mol. Med. 2020, 98, 1505–1509. [Google Scholar] [CrossRef]
- Delanghe, J.R.; Speeckaert, M.M.; De Buyzere, M.L. COVID-19 infections are also affected by human ACE1 D/I polymorphism. Clin. Chem. Lab. Med. 2020, 58, 1125–1126. [Google Scholar] [CrossRef] [Green Version]
- Cenanovic, M.; Dogan, S.; Asic, A.; Besic, L.; Marjanovic, D. Distribution of the ACE1 D Allele in the Bosnian-Herzegovinian Population and its Possible Role in the Regional Epidemiological Picture of COVID-19. Genet. Test. Mol. Biomarkers 2021, 25, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Annunziata, A.; Coppola, A.; Pafundi, P.C.; Guarino, S.; Di Spirito, V.; Maddaloni, V.; Pepe, N.; Fiorentino, G. ACE Gene I/D Polymorphism and Acute Pulmonary Embolism in COVID19 Pneumonia: A Potential Predisposing Role. Front. Med. 2021, 7, 631148. [Google Scholar] [CrossRef]
- Annunziata, A.; Coppola, A.; Lanza, M.; Simioli, F.; Imitazione, P.; Pepe, N.; Maddaloni, V.; Atripaldi, L.; Fiorentino, G. ACE DD polymorphism in severe COVID-19. J. Transl. Sci. 2021, 7. [Google Scholar] [CrossRef]
- Gómez, J.; Albaiceta, G.M.; García-Clemente, M.; López-Larrea, C.; Amado-Rodríguez, L.; Lopez-Alonso, I.; Hermida, T.; Enriquez, A.I.; Herrero, P.; Melón, S.; et al. Angiotensin-converting enzymes (ACE, ACE2) gene variants and COVID-19 outcome. Gene 2020, 762, 145102. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Abbas, M.; Verma, S.; Khan, F.H.; Raza, S.T.; Siddiqi, Z.; Ahmad, I.; Mahdi, F. Impact of I/D polymorphism of angiotensin-converting enzyme 1 (ACE1) gene on the severity of COVID-19 patients. Infect. Genet. Evol. 2021, 91, 104801. [Google Scholar] [CrossRef] [PubMed]
- Gunal, O.; Sezer, O.; Ustun, G.U.; Ozturk, C.E.; Sen, A.; Yigit, S.; Demirag, M.D. Angiotensin-converting enzyme-1 gene insertion/deletion polymorphism may be associated with COVID-19 clinical severity: A prospective cohort study. Ann. Saudi Med. 2021, 41, 141–146. [Google Scholar] [CrossRef]
- Hubacek, J.A.; Dusek, L.; Majek, O.; Adamek, V.; Cervinkova, T.; Dlouha, D.; Adamkova, V. ACE I/D polymorphism in Czech first-wave SARS-CoV-2-positive survivors. Clin. Chim. Acta 2021, 519, 206–209. [Google Scholar] [CrossRef]
- Jabotian, K. The Role of Angiotensin Converting Enzyme Insertion/Deletion Genetic Polymorphism in The Risk, Severity and Prognosis of COVID-19 Infection; The American University of Beirut: Beirut, Lebanon, 2021. [Google Scholar]
- Saab, Y.B.; Gard, P.R.; Overall, A.D. The geographic distribution of the ACE II genotype: A novel finding. Genet. Res. 2007, 89, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, R.; Morris, A.D.; Struthers, A.D. Angiotensin-converting enzyme gene polymorphism and cardiovascular disease. Clin. Sci. 1997, 93, 391–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasler, J.; Samuelsson, T.; Strub, K. Useful ‘junk’: Alu RNAs in the human transcriptome. Cell Mol. Life Sci. 2007, 64, 1793–1800. [Google Scholar] [CrossRef] [Green Version]
- Kazazian, H.H., Jr.; Moran, J.V. Mobile DNA in Health and Disease. N. Engl. J. Med. 2017, 377, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Payer, L.M.; Steranka, J.P.; Ardeljan, D.; Walker, J.; Fitzgerald, K.C.; Calabresi, P.A.; Cooper, T.A.; Burns, K.H. Alu insertion variants alter mRNA splicing. Nucleic Acids Res. 2019, 47, 421–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, D.; Rinn, J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 2012, 13, R107. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Schifanella, L.; Larsen, P.A. Alu retrotransposons and COVID-19 susceptibility and morbidity. Hum. Genom. 2021, 15, 2. [Google Scholar] [CrossRef] [PubMed]
- Mafra, F.F.P.; Gattai, P.P.; Macedo, M.M.; Mori, M.A.; Araujo, R.C. The angiotensin-I-converting enzyme insertion/deletion in polymorphic element codes for an AluYa5 RNA that downregulates gene expression. Pharm. J. 2018, 18, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Mariner, P.D.; Walters, R.D.; Espinoza, C.A.; Drullinger, L.F.; Wagner, S.D.; Kugel, J.F.; Goodrich, J.A. Human Alu RNA Is a Modular Transacting Repressor of mRNA Transcription during Heat Shock. Mol. Cell 2008, 29, 499–509. [Google Scholar] [CrossRef]
- Rigat, B.; Hubert, C.; Alhenc-Gelas, F.; Cambien, F.; Corvol, P.; Soubrier, F. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J. Clin. Investig. 1990, 86, 1343–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, Y.; Dai, C.; Wang, K.; Yu, J.; Tan, Y.; Zhang, W.; Yu, X.-F. Characterization of the relationship between APOBEC3B deletion and ACE Alu insertion. PLoS ONE 2013, 8, e64809. [Google Scholar] [CrossRef] [Green Version]
- Bogerd, H.P.; Wiegand, H.L.; Doehle, B.P.; Cullen, B.R. The intrinsic antiretroviral factor APOBEC3B contains two enzymatically active cytidine deaminase domains. Virology 2007, 364, 486–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rösler, C.; Köck, J.; Kann, M.; Malim, M.H.; Blum, H.E.; Baumert, T.F.; von Weizsäcker, F. APOBEC-mediated interference with hepadnavirus production. Hepatology 2005, 42, 301–309. [Google Scholar] [CrossRef]
- Hanscombe, K.B.; Traylor, M.; Hysi, P.G.; Bevan, S.; Dichgans, M.; Rothwell, P.M.; Worrall, B.B.; Seshadri, S.; Sudlow, C.; Williams, F.; et al. Genetic Factors Influencing Coagulation Factor XIII B-Subunit Contribute to Risk of Ischemic Stroke. Stroke 2015, 46, 2069–2074. [Google Scholar] [CrossRef]
- Bereczky, Z.; Muszbek, L. Factor XIII and venous thromboembolism. Semin. Thromb. Hemost. 2011, 37, 305–314. [Google Scholar] [CrossRef]
- Mezei, Z.A.; Bereczky, Z.; Katona, É.; Gindele, R.; Balogh, E.; Fiatal, S.; Balogh, L.; Czuriga, I.; Ádány, R.; Édes, I.; et al. Factor XIII B Subunit Polymorphisms and the Risk of Coronary Artery Disease. Int. J. Mol. Sci. 2015, 16, 1143–1159. [Google Scholar] [CrossRef]
- Matsuyama, T.; Kubli, S.P.; Yoshinaga, S.K.; Pfeffer, K.; Mak, T.W. An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 2020, 27, 3209–3225. [Google Scholar] [CrossRef]
- Ichinose, A. Factor XIII is a key molecule at the intersection of coagulation and fibrinolysis as well as inflammation and infection control. Int. J. Hematol. 2012, 95, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.R.; Jang, Y.; Kim Yoon, S.; Park, J.K.; Sorn, S.R.; Park, M.-Y.; Lee, M. The Impact of CDH13 Polymorphism and Statin Administration on TG/HDL Ratio in Cardiovascular Patients. Yonsei Med. J. 2015, 56, 1604–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas-Alarcon, G.; Martinez-Rodriguez, N.; Velazquez-Cruz, R.; Perez-Mendez, O.; Posadas-Sanchez, R.; Posadas-Romero, C.; Peña-Duque, M.A.; Martinez-Rios, M.A.; Ramirez-Fuentes, S.; Fragoso, J.M. The T > A (rs11646213) gene polymorphism of cadherin-13 ( CDH13 ) gene is associated with decreased risk of developing hypertension in Mexican population. Immunobiology 2017, 222, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, D.; Schoenenberger, A.W.; Dasen, B.; Erne, P.; Resink, T.J.; Philippova, M. Plasma T-cadherin negatively associates with coronary lesion severity and acute coronary syndrome. Eur. Heart J. Acute Cardiovasc. Care 2015, 4, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Delvoux, B.; Fischer, D.C.; Groothuis, P. The PROGINS polymorphism of the human progesterone receptor diminishes the response to progesterone. J. Mol. Endocrinol. 2007, 38, 331–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margaglione, M.; Grandone, E.; Vecchione, G.; Cappucci, G.; Giuliani, N.; Colaizzo, D.; Celentano, E.; Panico, S.; Di Minno, G. Plasminogen Activator Inhibitor-1 (PAI-1) Antigen Plasma Levels in Subjects Attending a Metabolic Ward: Relation to Polymorphisms of PAI-1 and Angiontensin Converting Enzyme (ACE) Genes. Arter. Thromb. Vasc. Biol. 1997, 17, 2082–2087. [Google Scholar] [CrossRef] [PubMed]
- Kerins, D.M.; Hao, Q.; Vaughan, D.E. Angiotensin induction of PAI-1 expression in endothelial cells is mediated by the hexapeptide angiotensin IV. J. Clin. Investig. 1995, 96, 2515–2520. [Google Scholar] [CrossRef] [Green Version]
- Whyte, C.S.; Morrow, G.B.; Mitchell, J.L.; Chowdary, P.; Mutch, N.J. Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat COVID-19. J. Thromb. Haemost. 2020, 18, 1548–1555. [Google Scholar] [CrossRef]
- Crouch, D.J.M.; Bodmer, W.F. Polygenic inheritance, GWAS, polygenic risk scores, and the search for functional variants. Proc. Natl. Acad. Sci. USA 2020, 117, 18924–18933. [Google Scholar] [CrossRef]
- Dai, S.-H.; Li, J.-F.; Feng, J.-B.; Li, R.-J.; Li, C.-B.; Li, Z.; Zhang, Y.; Li, D.-Q. Association of serum levels of AngII, KLK1, and ACE/KLK1 polymorphisms with acute myocardial infarction induced by coronary artery stenosis. J. Renin. Angiotensin Aldosterone Syst. 2016, 17, 1470320316655037. [Google Scholar] [CrossRef] [Green Version]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef]
- Fenner, F.; White, D.O. Medical Virology, 2nd ed.; Academic Press: New York, NY, USA, 1976; Volume XVII, p. 487. [Google Scholar]
- Gelfand, M.J.; Jackson, J.C.; Pan, X.; Nau, D.; Pieper, D.; Denison, E.; Dagher, M.; Lange, P.A.M.V.; Chiu, C.-Y.; Wang, M. The relationship between cultural tightness–looseness and COVID-19 cases and deaths: A global analysis. Lancet Planet. Health 2021, 5, e135–e144. [Google Scholar] [CrossRef]
- Yamamoto, N.; Bauer, G. Apparent difference in fatalities between Central Europe and East Asia due to SARS-COV-2 and COVID-19: Four hypotheses for possible explanation. Med. Hypotheses 2020, 144, 110160. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Country | Reference | ACE1 I/D Genotype | Control | COVID-19 Patients | Statistics | ||||
|---|---|---|---|---|---|---|---|---|---|
| Italy | Calabrese, 2020 [47] | Control (n = 111) | PE- (n = 43) | PE+ (n = 25) | p | ||||
| II | 13 (11.7%) | 2 (4.7%) | 3 (12%) | ||||||
| ID | 50 (45%) | 21 (48.8%) | 4 (16%) | ||||||
| DD | 48 (43.3%) | 20 (46.5%) | 18 (72%) | 0.03 | |||||
| Italy | Annunziata, 2020 [48] | Patients (n = 27) | PaO2/FiO2 (mmHg) | ||||||
| II | 2 (8%) | >200 | |||||||
| ID | 6 (23%) | 86.9 ± 15.3 | |||||||
| DD | 19 (73%) | 75.6 ± 11.3 | |||||||
| Spain | Gomez, 20201 [49] | Control (n = 248) | COVID (n = 125) | p | |||||
| II | 40 (16%) | 6 (5%) | |||||||
| ID | 123 (50%) | 66 (53%) | |||||||
| DD | 85 (34%) | 53 (42%) | 0.13 | ||||||
| Mild (n = 72) | Severe (n = 53) | p | |||||||
| II | 4(5%) | 2 (4%) | |||||||
| ID | 43(60%) | 23 (43%) | |||||||
| DD | 25(35%) | 28 (53%) | 0.04 | ||||||
| India | Verma, 2021 [50] | Mild (n = 149) | Severe (n = 120) | OR (p-value) | |||||
| II | 74 (49.7%) | 42 (35.0%) | 1 (ref) | ||||||
| ID | 58 (38.9%) | 48 (40.0%) | 1.54 (p = 0.17) | ||||||
| DD | 17 (11.4%) | 30 (25.0%) | 3.69 (p = 0.002) | ||||||
| Turkey | Gunal, 2021 [51] | Asymptomatic (n = 30) | Mild (n = 30) | Severe (n = 30) | p | ||||
| II | 15 (50%) | 7 (23.3%) | 9 (30%) | 0.08 | |||||
| ID | 4 (13.3%) | 8 (26.7%) | 2 (6.7%) | 0.09 | |||||
| DD | 11 (36.7%) | 15 (50%) | 19 (63.3%) | 0.12 | |||||
| Czech Republic | Hubacek, 2021 [52] | Control (n = 2579) | Asymptomatic (n = 163) | Symptomatic (n = 245) | OR Cont. vs. Asympt.(p-value) | OR Cont. vs. Sympt.(p-value) | |||
| II | 547 (21.2%) | 36 (22.1%) | 71 (29.0%) | 1.15 (p = 0.55) | 1.78 (p = 0.002) | ||||
| ID | 1331 (51.6%) | 87 (53.4%) | 123 (50.2%) | 1.14 (p = 0.49) | 1.27 (p = 0.16) | ||||
| DD | 701 (27.2%) | 40 (24.5%) | 51 (20.8%) | 1 (ref) | 1 (ref) | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, N.; Nishida, N.; Yamamoto, R.; Gojobori, T.; Shimotohno, K.; Mizokami, M.; Ariumi, Y. Angiotensin–Converting Enzyme (ACE) 1 Gene Polymorphism and Phenotypic Expression of COVID-19 Symptoms. Genes 2021, 12, 1572. https://doi.org/10.3390/genes12101572
Yamamoto N, Nishida N, Yamamoto R, Gojobori T, Shimotohno K, Mizokami M, Ariumi Y. Angiotensin–Converting Enzyme (ACE) 1 Gene Polymorphism and Phenotypic Expression of COVID-19 Symptoms. Genes. 2021; 12(10):1572. https://doi.org/10.3390/genes12101572
Chicago/Turabian StyleYamamoto, Naoki, Nao Nishida, Rain Yamamoto, Takashi Gojobori, Kunitada Shimotohno, Masashi Mizokami, and Yasuo Ariumi. 2021. "Angiotensin–Converting Enzyme (ACE) 1 Gene Polymorphism and Phenotypic Expression of COVID-19 Symptoms" Genes 12, no. 10: 1572. https://doi.org/10.3390/genes12101572