
Phenoconversion from Spastic Paraplegia to ALS/FTD Associated with CYP7B1 Compound Heterozygous Mutations

 ,
,  , ,
, ,
Abstract
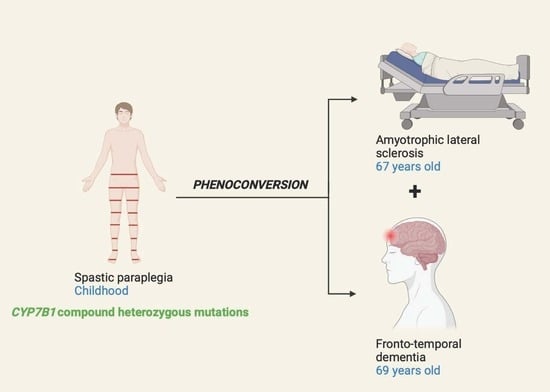
:
{kind=link}
{kind=link}
1. Introduction
2. Case Report
2.1. Proband
2.2. Parents
2.3. Molecular Genetic Analyses
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schüle, R.; Wiethoff, S.; Martus, P.; Karle, K.N.; Otto, S.; Klebe, S.; Klimpe, S.; Gallenmüller, C.; Kurzwelly, D.; Henkel, D.; et al. Hereditary Spastic Paraplegia: Clinicogenetic Lessons from 608 Patients: Hereditary Spastic Paraplegia. Ann. Neurol. 2016, 79, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Schöls, L.; Rattay, T.W.; Martus, P.; Meisner, C.; Baets, J.; Fischer, I.; Jägle, C.; Fraidakis, M.J.; Martinuzzi, A.; Saute, J.A.; et al. Hereditary Spastic Paraplegia Type 5: Natural History, Biomarkers and a Randomized Controlled Trial. Brain 2017, 140, 3112–3127. [Google Scholar] [CrossRef] [PubMed]
- Marelli, C.; Lamari, F.; Rainteau, D.; Lafourcade, A.; Banneau, G.; Humbert, L.; Monin, M.-L.; Petit, E.; Debs, R.; Castelnovo, G.; et al. Plasma oxysterols: Biomarkers for diagnosis and treatment in spastic paraplegia type 5. Brain 2017, 141, 72–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manganelli, F.; Pisciotta, C.; Dubbioso, R.; Iodice, R.; Criscuolo, C.; Ruggiero, L.; Michele, G.D.; Santoro, L. Electrophysiological Characterisation in Hereditary Spastic Paraplegia Type 5. Clin. Neurophysiol. 2011, 122, 819–822. [Google Scholar] [CrossRef] [PubMed]
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Nicolas, A.; Kenna, K.P.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-Wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teyssou, E.; Chartier, L.; Amador, M.-D.-M.; Lam, R.; Lautrette, G.; Nicol, M.; Machat, S.; Da Barroca, S.; Moigneu, C.; Mairey, M.; et al. Novel UBQLN2 Mutations Linked to Amyotrophic Lateral Sclerosis and Atypical Hereditary Spastic Paraplegia Phenotype through Defective HSP70-Mediated Proteolysis. Neurobiol. Aging 2017, 58, 239.e11–239.e20. [Google Scholar] [CrossRef] [PubMed]
- Amador, M.-D.-M.; Muratet, F.; Teyssou, E.; Banneau, G.; Danel-Brunaud, V.; Allart, E.; Antoine, J.-C.; Camdessanché, J.-P.; Anheim, M.; Rudolf, G.; et al. Spastic Paraplegia Due to Recessive or Dominant Mutations in ERLIN2 Can Convert to ALS. Neurol Genet 2019, 5, e374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strong, M.J.; Gordon, P.H. Primary lateral sclerosis, hereditary spastic paraplegia and amyotrophic lateral sclerosis: Discrete entities or spectrum? Amyotroph. Lateral Scler. 2005, 6, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Shefner, J.M.; Al-Chalabi, A.; Baker, M.R.; Cui, L.-Y.; de Carvalho, M.; Eisen, A.; Grosskreutz, J.; Hardiman, O.; Henderson, R.; Matamala, J.M.; et al. A Proposal for New Diagnostic Criteria for ALS. Clin. Neurophysiol. 2020, 131, 1975–1978. [Google Scholar] [CrossRef] [PubMed]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; van Swieten, J.C.; Seelaar, H.; Dopper, E.G.P.; Onyike, C.U.; et al. Sensitivity of Revised Diagnostic Criteria for the Behavioural Variant of Frontotemporal Dementia. Brain 2011, 134, 2456–2477. [Google Scholar] [CrossRef] [PubMed]
- Goizet, C.; Boukhris, A.; Durr, A.; Beetz, C.; Truchetto, J.; Tesson, C.; Tsaousidou, M.; Forlani, S.; Guyant-Marechal, L.; Fontaine, B.; et al. CYP7B1 Mutations in Pure and Complex Forms of Hereditary Spastic Paraplegia Type 5. Brain 2009, 132, 1589–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.D.; Vesely, P.W.; Datta, K.; Gerace, L. Erlins Restrict SREBP Activation in the ER and Regulate Cellular Cholesterol Homeostasis. J. Cell Biol. 2013, 203, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, H.; Ho, W.Y.; Chang, J.; Ling, S. Cholesterol Dyshomeostasis in Amyotrophic Lateral Sclerosis: Cause, Consequence, or Epiphenomenon? FEBS J. 2021, 16175. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-M.; Noh, M.-Y.; Kim, H.; Cheon, S.-Y.; Lee, K.M.; Lee, J.; Cha, E.; Park, K.S.; Lee, K.-W.; Sung, J.-J.; et al. 25-hydroxycholesterol is involved in the pathogenesis of amyotrophic lateral sclerosis. Oncotarget 2017, 8, 11855–11867. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Theuriet, J.; Pegat, A.; Leblanc, P.; Vukusic, S.; Cazeneuve, C.; Millecamps, S.; Banneau, G.; Guillaud-Bataille, M.; Bernard, E. Phenoconversion from Spastic Paraplegia to ALS/FTD Associated with CYP7B1 Compound Heterozygous Mutations. Genes 2021, 12, 1876. https://doi.org/10.3390/genes12121876
Theuriet J, Pegat A, Leblanc P, Vukusic S, Cazeneuve C, Millecamps S, Banneau G, Guillaud-Bataille M, Bernard E. Phenoconversion from Spastic Paraplegia to ALS/FTD Associated with CYP7B1 Compound Heterozygous Mutations. Genes. 2021; 12(12):1876. https://doi.org/10.3390/genes12121876
Chicago/Turabian StyleTheuriet, Julian, Antoine Pegat, Pascal Leblanc, Sandra Vukusic, Cécile Cazeneuve, Stéphanie Millecamps, Guillaume Banneau, Marine Guillaud-Bataille, and Emilien Bernard. 2021. "Phenoconversion from Spastic Paraplegia to ALS/FTD Associated with CYP7B1 Compound Heterozygous Mutations" Genes 12, no. 12: 1876. https://doi.org/10.3390/genes12121876
APA StyleTheuriet, J., Pegat, A., Leblanc, P., Vukusic, S., Cazeneuve, C., Millecamps, S., Banneau, G., Guillaud-Bataille, M., & Bernard, E. (2021). Phenoconversion from Spastic Paraplegia to ALS/FTD Associated with CYP7B1 Compound Heterozygous Mutations. Genes, 12(12), 1876. https://doi.org/10.3390/genes12121876