
Adducted Thumb and Peripheral Polyneuropathy: Diagnostic Supports in Suspecting White–Sutton Syndrome: Case Report and Review of the Literature

 , , , , ,
, , , , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
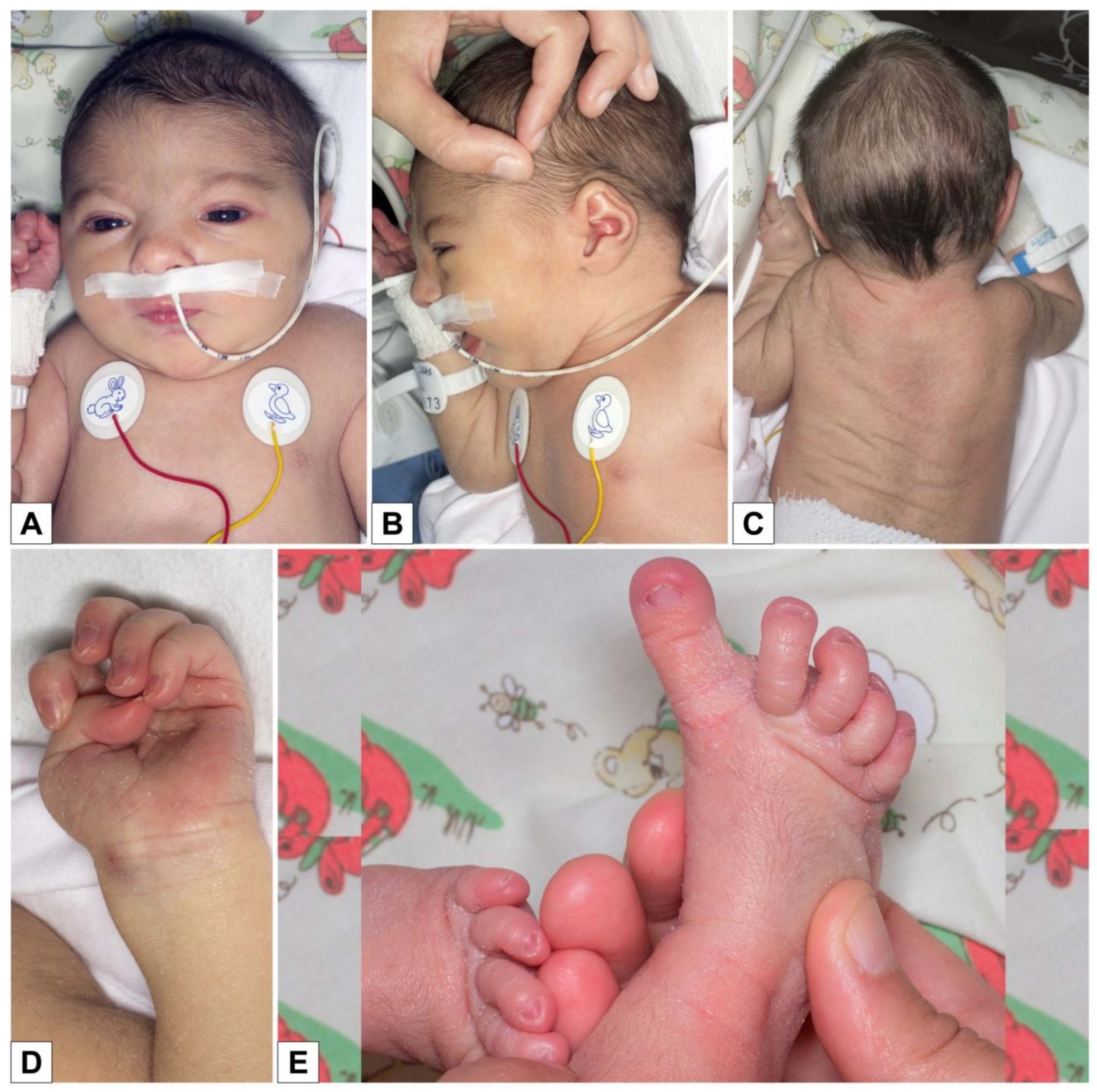
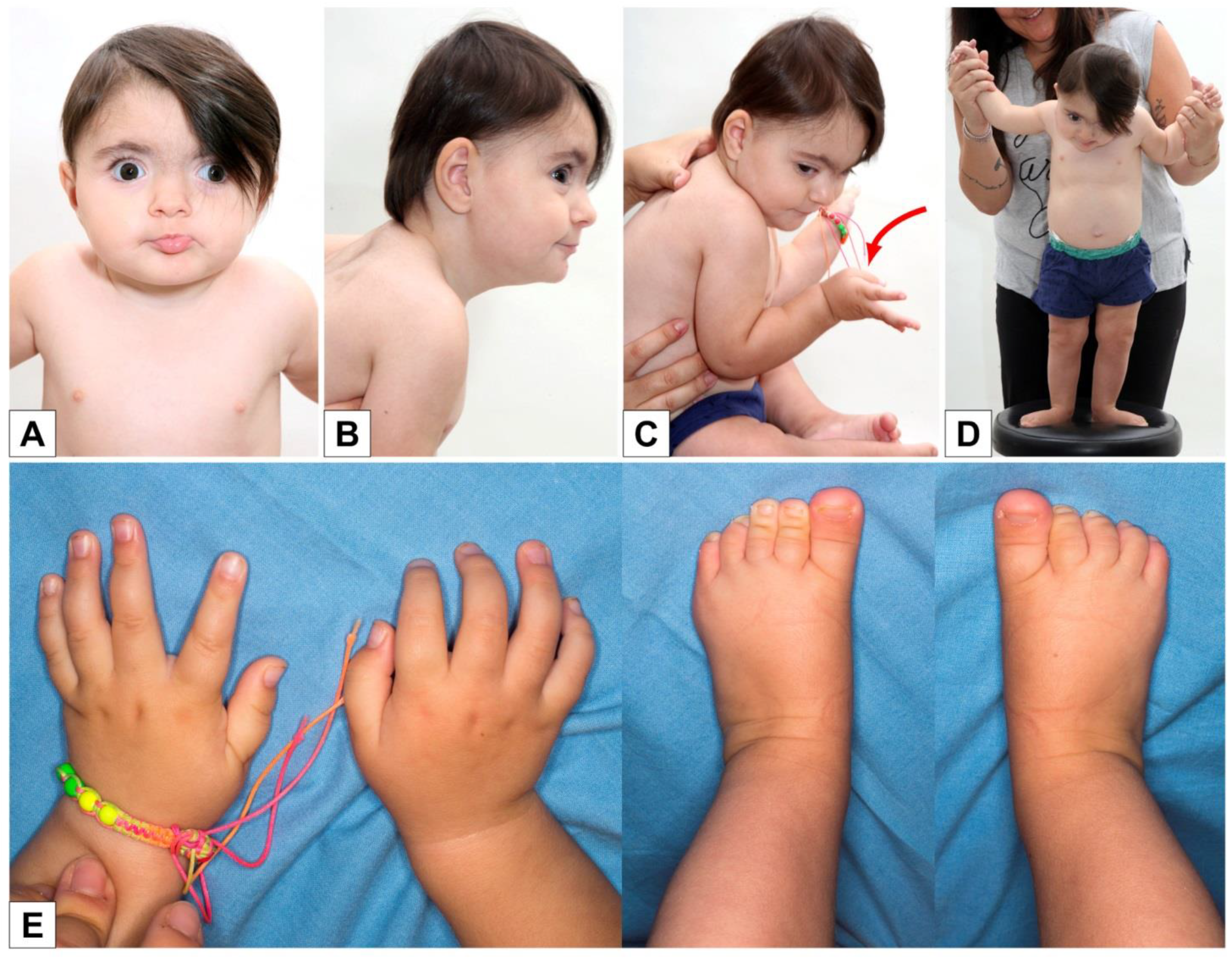
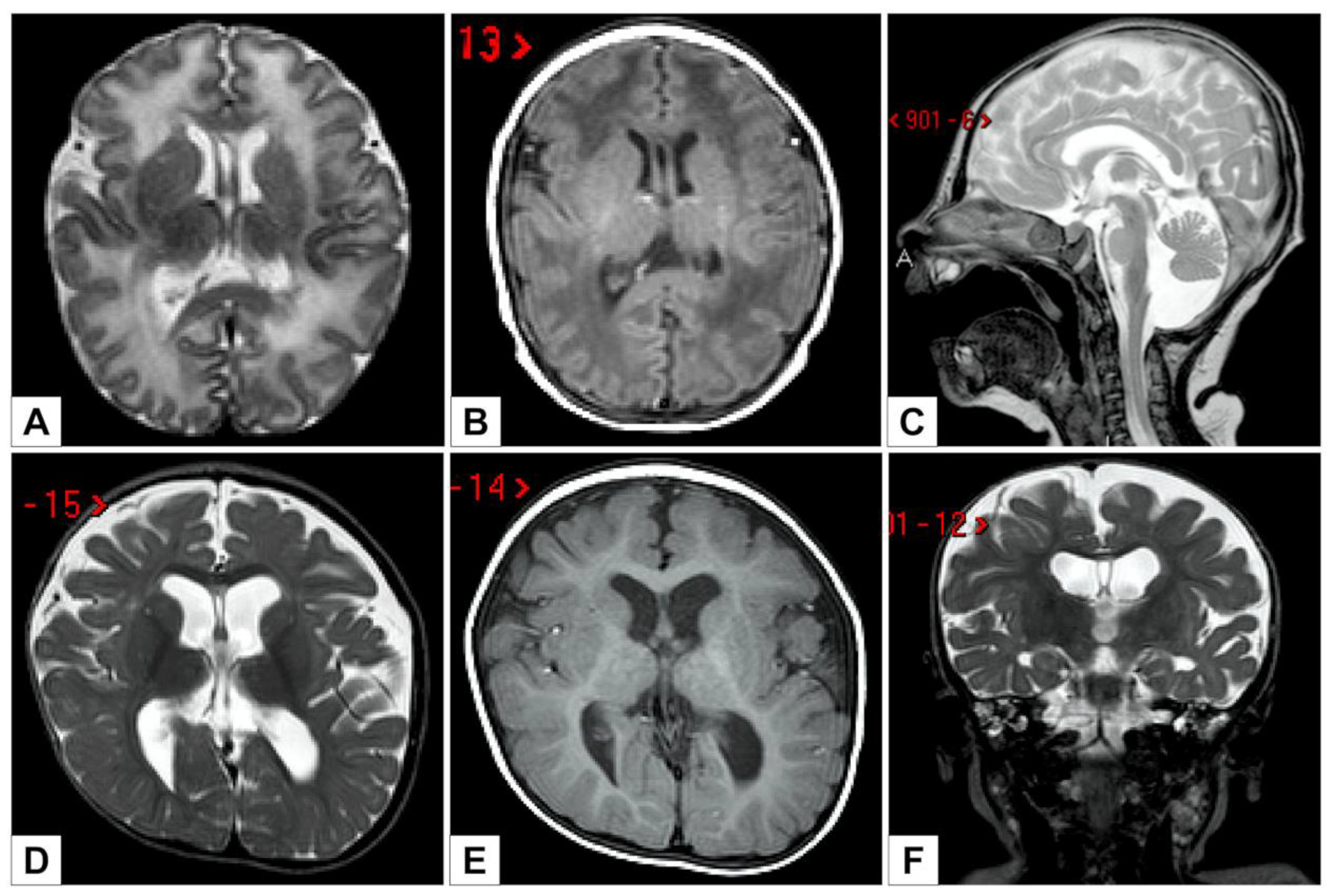
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ye, Y.; Cho, M.T.; Retterer, K.; Alexander, N.; Ben-Omran, T.; Al-Mureikhi, M.; Cristian, I.; Wheeler, P.G.; Crain, C.; Zand, D.J.; et al. De novoPOGZmutations are associated with neurodevelopmental disorders and microcephaly. Mol. Case Stud. 2015, 1, a000455. [Google Scholar] [CrossRef] [Green Version]
- White, J.; Beck, C.R.; Harel, T.; Posey, J.E.; Jhangiani, S.N.; Tang, S.; Farwell, K.D.; Powis, Z.; Mendelsohn, N.J.; Baker, J.A.; et al. POGZ truncating alleles cause syndromic intellectual disability. Genome Med. 2016, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, R.; Nakazawa, T.; Tsurusaki, Y.; Yasuda, Y.; Nagayasu, K.; Matsumura, K.; Kawashima, H.; Yamamori, H.; Fujimoto, M.; Ohi, K.; et al. Whole-exome sequencing and neurite outgrowth analysis in autism spectrum disorder. J. Hum. Genet. 2015, 61, 199–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stessman, H.A.; Willemsen, M.H.; Fenckova, M.; Penn, O.; Hoischen, A.; Xiong, B.; Wang, T.; Hoekzema, K.; Vives, L.; Vogel, I.; et al. Disruption of POGZ Is Associated with Intellectual Disability and Autism Spectrum Disorders. Am. J. Hum. Genet. 2016, 98, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, K.; Nakazawa, T.; Nagayasu, K.; Gotoda-Nishimura, N.; Kasai, A.; Hayata-Takano, A.; Shintani, N.; Yamamori, H.; Yasuda, Y.; Hashimoto, R.; et al. De novo POGZ mutations in sporadic autism disrupt the DNA-binding activity of POGZ. J. Mol. Psychiatry 2016, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.; Zou, Y.; Zhang, Y.; Zhang, R.; Ou, J.; Shen, Y.; Zhao, J.; Luo, X.; Guo, J.; Zeng, L.; et al. A novel de novo POGZ mutation in a patient with intellectual disability. J. Hum. Genet. 2016, 61, 357–359. [Google Scholar] [CrossRef]
- Dentici, M.L.; Niceta, M.; Pantaleoni, F.; Barresi, S.; Bencivenga, P.; Dallapiccola, B.; Digilio, M.C.; Tartaglia, M. Expanding the phenotypic spectrum of truncating POGZ mutations: Association with CNS malformations, skeletal abnormalities, and distinctive facial dysmorphism. Am. J. Med. Genet. Part A 2017, 173, 1965–1969. [Google Scholar] [CrossRef]
- Ferretti, A.; Barresi, S.; Trivisano, M.; Ciolfi, A.; Dentici, M.L.; Radio, F.C.; Vigevano, F.; Tartaglia, M.; Specchio, N. POGZ-related epilepsy: Case report and review of the literature. Am. J. Med. Genet. Part A 2019, 179, 1631–1636. [Google Scholar] [CrossRef]
- Zhao, W.; Quan, Y.; Wu, H.; Han, L.; Bai, T.; Ma, L.; Li, B.; Xun, G.; Ou, J.; Zhao, J.; et al. POGZ de novo missense variants in neuropsychiatric disorders. Mol. Genet. Genom. Med. 2019, 7, e900. [Google Scholar] [CrossRef] [Green Version]
- Batzir, N.A.; Posey, J.E.; Song, X.; Akdemir, Z.C.; Rosenfeld, J.A.; Brown, C.W.; Chen, E.X.; Holtrop, S.G.; Mizerik, E.; Moreno, M.N.; et al. Phenotypic expansion of POGZ -related intellectual disability syndrome (White-Sutton syndrome). Am. J. Med. Genet. Part A 2020, 182, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Dal, S.; Hopper, B.; du Chattel, M.V.R.; Goel, H. A case of White–Sutton syndrome with previously described loss-of-function variant in DDE domain of POGZ (p.Arg1211*) and Kartagener syndrome. Am. J. Med Genet. Part A 2021, 185, 1006–1007. [Google Scholar] [CrossRef]
- Garde, A.; Cornaton, J.; Sorlin, A.; Moutton, S.; Nicolas, C.; Juif, C.; Geneviève, D.; Perrin, L.; Khau-Van-Kien, P.; Smol, T.; et al. Neuropsychological study in 19 French patients with White-Sutton syndrome and POGZ mutations. Clin. Genet. 2021, 99, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Pascolini, G.; Agolini, E.; Fleischer, N.; Gulotta, E.; Cesario, C.; D’Elia, G.; Novelli, A.; Majore, S.; Grammatico, P. A novel patient with White–Sutton syndrome refines the mutational and clinical repertoire of the POGZ- related phenotype and suggests further observations. Am. J. Med. Genet. Part A 2020, 182, 1791–1795. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Ramakrishnaiah, R.; Schaefer, B. The neurological aspects related to POGZ mutation: Case report and review of CNS malformations and epilepsy. Acta Neurol. Belg. 2019, 120, 447–450. [Google Scholar] [CrossRef]
- OMIM—Online Mendelian Inheritance in Man®. Available online: https://www.omim.org/ (accessed on 18 May 2021).
- Bauer, C.K.; Calligari, P.; Radio, F.C.; Caputo, V.; Dentici, M.L.; Falah, N.; High, F.; Pantaleoni, F.; Barresi, S.; Ciolfi, A.; et al. Mutations in KCNK4 that Affect Gating Cause a Recognizable Neurodevelopmental Syndrome. Am. J. Hum. Genet. 2018, 103, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Flex, E.; Martinelli, S.; Van Dijck, A.; Ciolfi, A.; Cecchetti, S.; Coluzzi, E.; Pannone, L.; Andreoli, C.; Radio, F.C.; Pizzi, S.; et al. Aberrant Function of the C-Terminal Tail of HIST1H1E Accelerates Cellular Senescence and Causes Premature Aging. Am. J. Hum. Genet. 2019, 105, 493–508. [Google Scholar] [CrossRef] [Green Version]
- Radio, F.C.; Pang, K.; Ciolfi, A.; Levy, M.A.; Hernández-García, A.; Pedace, L.; Pantaleoni, F.; Liu, Z.; de Boer, E.; Jackson, A.; et al. SPEN haploinsufficiency causes a neurodevelopmental disorder overlapping proximal 1p36 deletion syndrome with an episignature of X chromosomes in females. Am. J. Hum. Genet. 2021, 108, 502–516. [Google Scholar] [CrossRef]
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Cingolani, P.; Sladek, R.; Blanchette, M. BigDataScript: A scripting language for data pipelines. Bioinformatics 2015, 31, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP v2.0: A Database of Human Non-synonymous SNVs and Their Functional Predictions and Annotations. Hum. Mutat. 2013, 34, E2393–E2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O‘Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Human Splicing Finder 3.1. Available online: http://umd.be/Redirect.html (accessed on 18 May 2021).
- Berkeley Drosophila Genome Project. Available online: https://www.fruitfly.org/seq_tools/splice.html (accessed on 18 May 2021).
- NetGene2 Server. Available online: http://www.cbs.dtu.dk/services/NetGene2/ (accessed on 18 May 2021).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- The Human Phenotype Ontology (HPO). Available online: https://hpo.jax.org/app/tools/phenomizer (accessed on 18 May 2021).
- Kaepernick, L.; Legius, E.; Higgins, J.; Kapur, S. Clinical aspects of the MASA syndrome in a large family, including expressing females. Clin. Genet. 2008, 45, 181–185. [Google Scholar] [CrossRef]
- Vos, Y.J.; De Walle, H.E.K.; Bos, K.K.; Stegeman, J.A.; Berge, A.M.T.; Bruining, M.; Van Maarle, M.C.; Elting, M.W.; Hollander, N.S.D.; Hamel, B.; et al. Genotype-phenotype correlations in L1 syndrome: A guide for genetic counselling and mutation analysis. J. Med. Genet. 2009, 47, 169–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Tan, J.; Zhu, T.; Ou, J.; Li, Y.; Shen, L.; Wu, H.; Han, L.; Liu, Y.; Jia, X.; et al. Rare inherited missense variants of POGZ associate with autism risk and disrupt neuronal development. J. Genet. Genom. 2019, 46, 247–257. [Google Scholar] [CrossRef]
- Campo, C.; Da Silva Filho, M.I.; Weinhold, N.; Goldschmidt, H.; Hemminki, K.; Merz, M.; Försti, A. Genetic Susceptibility to Bortezomib-Induced Peripheral Neuroropathy: Replication of the Reported Candidate Susceptibility Loci. Neurochem. Res. 2016, 42, 925–931. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Clinical Feature | Frequency (%) | Clinical Feature | Frequency (%) |
|---|---|---|---|
| Developmental delay | 100 | GI manifestations | 61 |
| Intellectual disability | 100 | Microcephaly | 47 |
| Speech delay | 100 | Obesity/increased BMI | 39 |
| Motor delay | 97 | Recurrent infections | 35 |
| Dysmorphism | 95 | Hypotonia | 29 |
| Behavioral phenotype | 79 | Sleep disorders | 25 |
| Ocular findings | 79 | Sensorineural hearing loss | 24 |
| ASD | 67 | Others | <20 |
| Brain MRI anomalies | 65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trimarchi, G.; Caraffi, S.G.; Radio, F.C.; Barresi, S.; Contrò, G.; Pizzi, S.; Maini, I.; Pollazzon, M.; Fusco, C.; Sassi, S.; et al. Adducted Thumb and Peripheral Polyneuropathy: Diagnostic Supports in Suspecting White–Sutton Syndrome: Case Report and Review of the Literature. Genes 2021, 12, 950. https://doi.org/10.3390/genes12070950
Trimarchi G, Caraffi SG, Radio FC, Barresi S, Contrò G, Pizzi S, Maini I, Pollazzon M, Fusco C, Sassi S, et al. Adducted Thumb and Peripheral Polyneuropathy: Diagnostic Supports in Suspecting White–Sutton Syndrome: Case Report and Review of the Literature. Genes. 2021; 12(7):950. https://doi.org/10.3390/genes12070950
Chicago/Turabian StyleTrimarchi, Gabriele, Stefano Giuseppe Caraffi, Francesca Clementina Radio, Sabina Barresi, Gianluca Contrò, Simone Pizzi, Ilenia Maini, Marzia Pollazzon, Carlo Fusco, Silvia Sassi, and et al. 2021. "Adducted Thumb and Peripheral Polyneuropathy: Diagnostic Supports in Suspecting White–Sutton Syndrome: Case Report and Review of the Literature" Genes 12, no. 7: 950. https://doi.org/10.3390/genes12070950