
Whole Exome Sequencing Is the Minimal Technological Approach in Probands Born to Consanguineous Couples

 , , , , , ,
, , , , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
3. Patients and Results
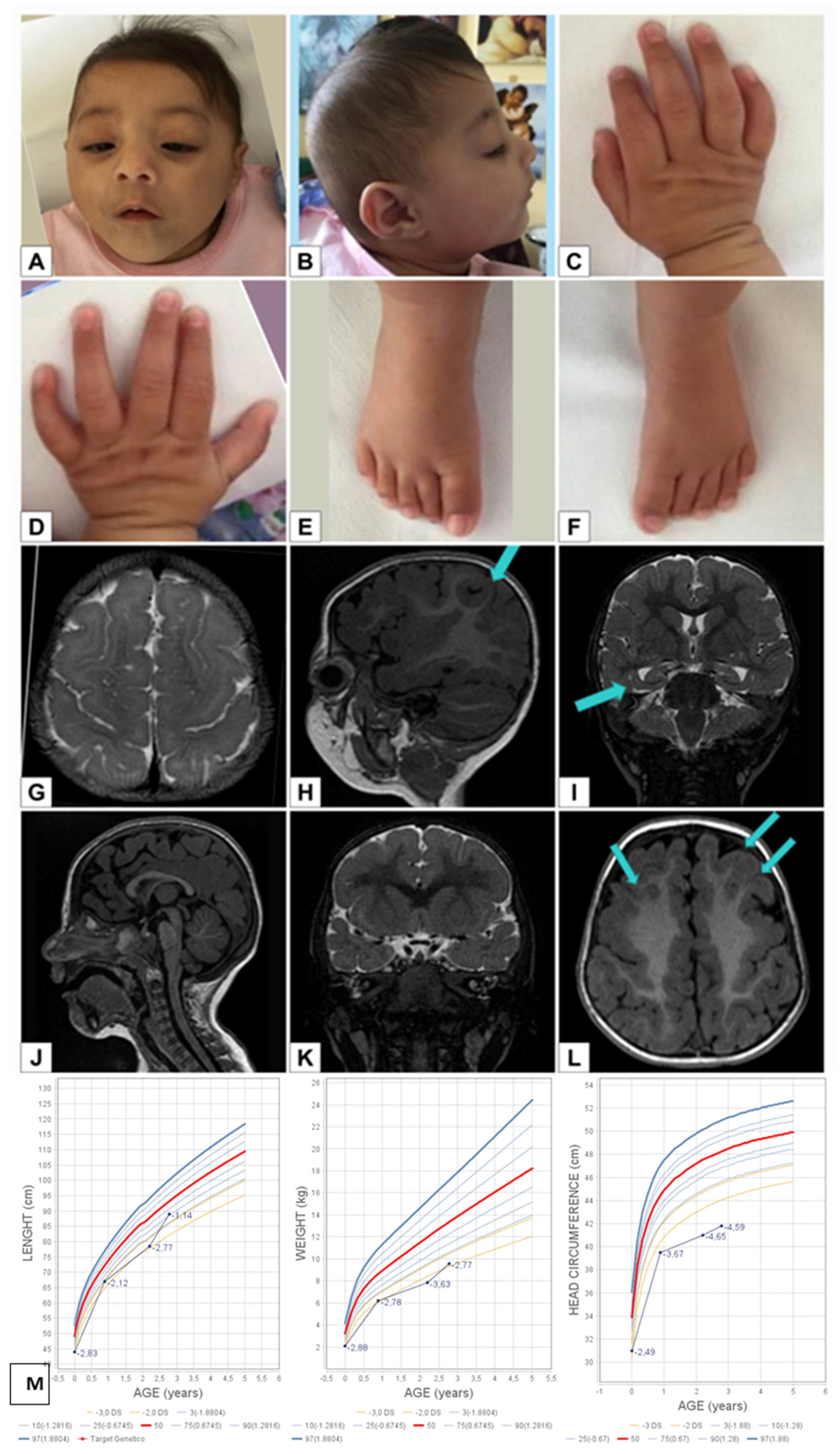
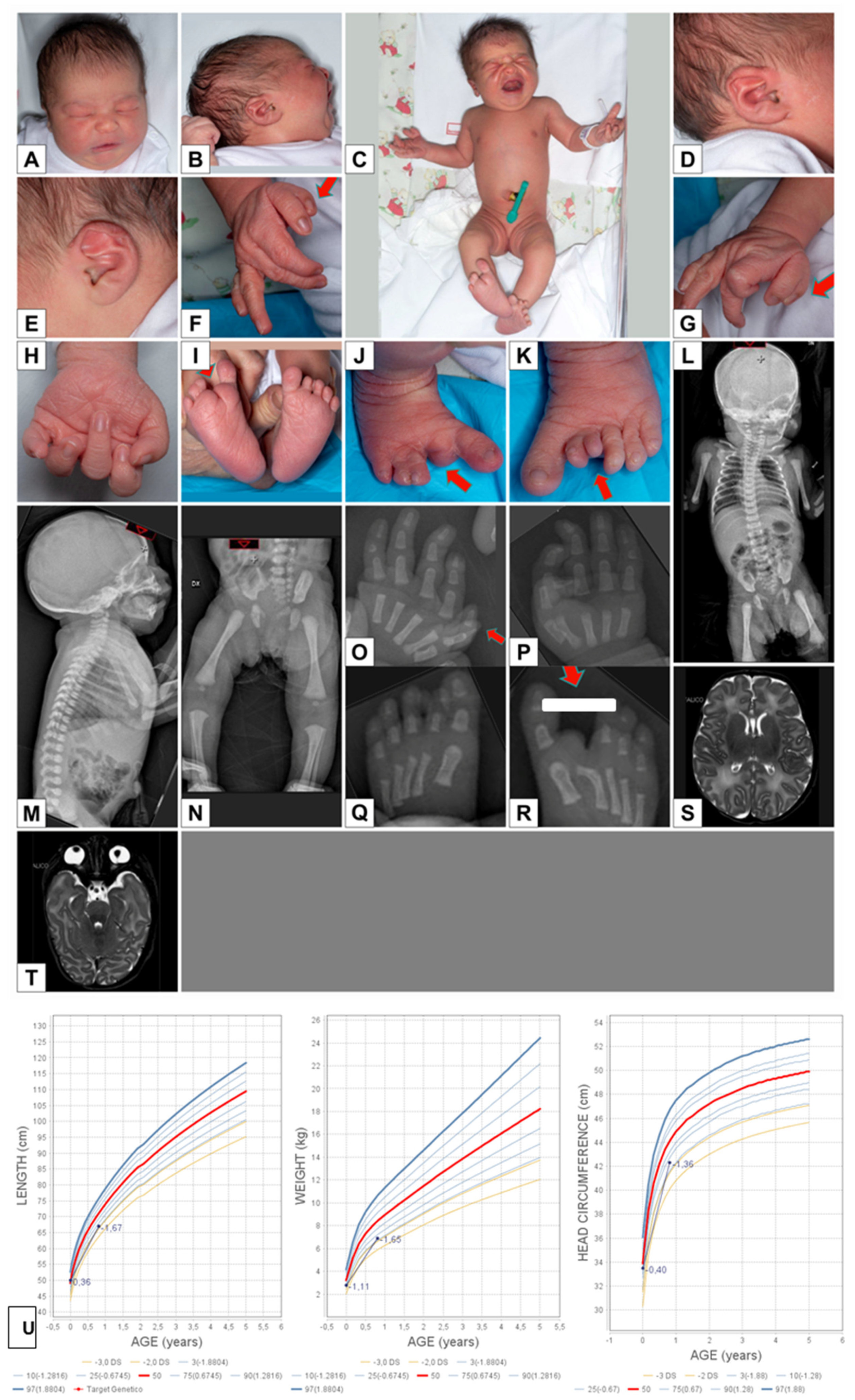
3.1. Case Reports
3.2. Case 1
3.3. Case 2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bittles, A.H.; Black, M.L. Evolution in health and medicine Sackler colloquium: Consanguinity, human evolution, and complex diseases. Proc. Natl. Acad. Sci. USA 2010, 107, 1779–1786. [Google Scholar] [CrossRef] [Green Version]
- Teeuw, M.; Loukili, G.; Bartels, E.; ten Kate, L.P.; Cornel, M.C.; Henneman, L. Consanguineous marriage and reproductive risk: Attitudes and understanding of ethnic groups practising consanguinity in western society. Eur. J. Hum. Genet. 2014, 22, 452–457. [Google Scholar] [CrossRef] [Green Version]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Teeuw, M.; Waisfisz, Q.; Zwijnenburg, P.J.; Sistermans, E.A.; Weiss, M.M.; Henneman, L.; ten Kate, L.P.; Cornel, M.C.; Meijers-Heijboer, H. First steps in exploring prospective exome sequencing of consanguineous couples. Eur. J. Med. Genet. 2014, 57, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, E.; Wright, J.; Corry, P.; Oddie, S.; Small, N.; Parslow, R.C. Analysis of the born in Bradford birth cohort–authors’ reply. Lancet 2014, 383, 123. [Google Scholar] [CrossRef]
- Komlosi, K.; Diederich, S.; Fend-Guella, D.L.; Bartsch, O.; Winter, J.; Zechner, U.; Beck, M.; Meyer, P.; Schweiger, S. Targeted next-generation sequencing analysis in couples at increased risk for autosomal recessive disorders. Orphanet J. Rare Dis. 2018, 13, 23. [Google Scholar] [CrossRef] [Green Version]
- Monies, D.; Abouelhoda, M.; Assoum, M.; Moghrabi, N.; Rafiullah, R.; Almontashiri, N.; Alowain, M.; Alzaidan, H.; Alsayed, M.; Subhani, S.; et al. Lessons learned from large-scale, first-tier clinical exome sequencing in a highly consanguineous population. Am. J. Hum. Genet. 2019, 105, 879. [Google Scholar] [CrossRef] [Green Version]
- Parrini, E.; Marini, C.; Mei, D.; Galuppi, A.; Cellini, E.; Pucatti, D.; Chiti, L.; Rutigliano, D.; Bianchini, C.; Virdò, S.; et al. Diagnostic targeted resequencing in 349 patients with drug-resistant pediatric epilepsies identifies causative mutations in 30 different genes. Hum. Mutat. 2017, 38, 216–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Oxf. Bioinform. 2018, bty897. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.F.; Pomp, O.; Ben-Omran, T.; Kodani, A.; Henke, K.; Mochida, G.H.; Yu, T.W.; Woodworth, M.B.; Bonnard, C.; Raj, G.S.; et al. Katanin p80 regulates human cortical development by limiting centriole and cilia number. Neuron 2014, 84, 1240–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra-Gorur, K.; Caglayan, A.O.; Schaffer, A.E.; Chabu, C.; Henegariu, O.; Vonhoff, F.; Akgumus, G.T.; Nishimura, S.; Han, W.; Tu, S.; et al. Mutations in KATNB1 cause complex cerebral malformations by disrupting asymmetrically dividing neural progenitors. Neuron 2014, 84, 1226–1239. [Google Scholar] [CrossRef]
- Ho, E.K.; Stearns, T. Hedgehog signaling and the primary cilium: Implications for spatial and temporal constraints on signaling. Development 2021, 148, dev195552. [Google Scholar] [CrossRef] [PubMed]
- Yigit, G.; Wieczorek, D.; Bogershausen, N.; Beleggia, F.; Moller-Hartmann, C.; Altmuller, J.; Thiele, H.; Nurnberg, P.; Wollnik, B. A syndrome of microcephaly, short stature, polysyndactyly, and dental anomalies caused by a homozygous KATNB1 mutation. Am. J. Med. Genet. Part A 2016, 170A, 728–733. [Google Scholar] [CrossRef]
- Lahrouchi, N.; George, A.; Ratbi, I.; Schneider, R.; Elalaoui, S.C.; Moosa, S.; Bharti, S.; Sharma, R.; Abu-Asab, M.; Onojafe, F.; et al. Homozygous frameshift mutations in FAT1 cause a syndrome characterized by colobomatous-microphthalmia, ptosis, nephropathy and syndactyly. Nat. Commun. 2019, 10, 1180. [Google Scholar] [CrossRef]
- Pastushenko, I.; Mauri, F.; Song, Y.; de Cock, F.; Meeusen, B.; Swedlund, B.; Impens, F.; Van Haver, D.; Opitz, M.; Thery, M.; et al. Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis. Nature 2021, 589, 448–455. [Google Scholar] [CrossRef]
- Gee, Y.H.; Sadowski, C.E.; Aggarwal, P.K.; Porath, J.D.; Yakulov, T.A.; Schueler, M.; Lovric, S.; Ashraf, S.; Braun, D.A.; Halbritter, J.; et al. FAT1 mutations cause a glomerulotubular nephropathy. Nat. Commun. 2016, 7, 10822. [Google Scholar] [CrossRef] [Green Version]
- Landini, S.; Mazzinghi, B.; Becherucci, F.; Allinovi, M.; Provenzano, A.; Palazzo, V.; Ravaglia, F.; Artuso, R.; Bosi, E.; Stagi, S.; et al. Reverse phenotyping after whole-exome sequencing in steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 2020, 15, 89–100. [Google Scholar] [CrossRef]
- Rossanti, R.; Watanabe, T.; Nagano, C.; Hara, S.; Horinouchi, T.; Yamamura, T.; Sakakibara, N.; Ninchoji, T.; Iijima, K.; Nozu, K. FAT1 biallelic truncating mutation causes a non-syndromic proteinuria in a child. CEN Case Rep. 2021, 10, 100–105. [Google Scholar] [CrossRef]
- Haug, P.; Koller, S.; Maggi, J.; Lang, E.; Feil, S.; Wlodarczyk, A.; Bähr, L.; Steindl, K.; Rohrbach, M.; Gerth-Kahlert, C.; et al. Whole exome sequencing in coloboma/ microphthalmia: Identification of novel and recurrent variants in seven genes. Genes 2021, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Gros, J.; Tabin, C.J. Vertebrate limb bud formation is initiated by localized epithelial-to-mesenchymal transition. Science 2014, 343, 1253–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussion, S.; Escande, F.; Jourdain, A.S.; Smol, T.; Brunelle, P.; Duhamel, C.; Alembik, Y.; Attié-Bitach, T.; Baujat, G.; Bazin, A.; et al. TAR syndrome: Clinical and molecular characterization of a cohort of 26 patients and description of novel noncoding variants of RBM8A. Hum. Mutat. 2020, 1220–1225. [Google Scholar] [CrossRef]
- Puppo, F.; Dionnet, E.; Gaillard, M.-C.; Gaildrat, P.; Castro, C.; Vovan, C.; Bertaux, K.; Bernard, R.; Attarian, S.; Goto, K.; et al. Identification of variants in the 4q35 Gene FAT1 in patients with a facioscapulohumeral dystrophy-like phenotype. Hum. Mutat. 2015, 36, 443–453. [Google Scholar] [CrossRef]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Oegema, R.; Barkovich, A.J.; Mancini, G.M.S.; Guerrini, R.; Dobyns, W.B. Subcortical heterotopic gray matter brain malformations: Classification study of 107 individuals. Neurology 2019, 93, e1360–e1373. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| CASE 1—Homozygous c.1416 + 1del in KATNB1 | CASE 2—Homozygous c.9729del in FAT1 | |||
|---|---|---|---|---|
| HPO Number | Phenotype | HPO Number | Phenotype | |
| HEAD | HP:0000252 | Microcephaly | ||
| HP:0011327 | Posterior plagiocephaly | |||
| MRI ANOMALIES | HP:0012766 | Widened cerebral subarachnoid space | ||
| HP:0002536 | Abnormal cortical gyration | |||
| HP:0025100 | Abnormal hippocampus morphology | |||
| HP:0032391 | Subcortical heterotopia | |||
| HP:0006891 | Thick cerebral cortex | |||
| EYES | HP:0000666 | Horizontal nystagmus | ||
| HP:0007703 | Abnormality of retinal pigmentation | |||
| HP:0001488 | Bilateral ptosis | HP:0001488 | Bilateral ptosis | |
| HP:0007968 | Remnants of the hyaloid vascular system | |||
| HP:0007633 | Bilateral microphthalmos | |||
| HP:0000588 | Optic nerve coloboma | |||
| HEART | HP:0011681 | Subarterial ventricular septal defect | HP:0011681 | Subarterial ventricular septal defect |
| HP:0001684 | Secundum atrial septal defect | HP:0001684 | Secundum atrial septal defect | |
| SKELETON | HP:0001177 | Preaxial hand polydactyly | ||
| HP:0001839 | Split foot | |||
| HP:0001829 | Foot polydactyly | |||
| HP:0004209 | Clinodactyly of the 5th finger | |||
| HP:0005709 | 2–3 toe cutaneous syndactyly | |||
| DEVELOPMENT AND GROWTH | ||||
| HP:0001510 | Growth delay | |||
| HP:0001263 | Global developmental delay | |||
| KIDNEY | ||||
| HP:0000100 | Nephrotic syndrome | |||
| HEARING | HP:0012716 | Moderate conductive hearing impairment | ||
| HP:0012712 | Mild hearing impairment | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peluso, F.; Caraffi, S.G.; Zuntini, R.; Trimarchi, G.; Ivanovski, I.; Valeri, L.; Barbieri, V.; Marinelli, M.; Pancaldi, A.; Melli, N.; et al. Whole Exome Sequencing Is the Minimal Technological Approach in Probands Born to Consanguineous Couples. Genes 2021, 12, 962. https://doi.org/10.3390/genes12070962
Peluso F, Caraffi SG, Zuntini R, Trimarchi G, Ivanovski I, Valeri L, Barbieri V, Marinelli M, Pancaldi A, Melli N, et al. Whole Exome Sequencing Is the Minimal Technological Approach in Probands Born to Consanguineous Couples. Genes. 2021; 12(7):962. https://doi.org/10.3390/genes12070962
Chicago/Turabian StylePeluso, Francesca, Stefano Giuseppe Caraffi, Roberta Zuntini, Gabriele Trimarchi, Ivan Ivanovski, Lara Valeri, Veronica Barbieri, Maria Marinelli, Alessia Pancaldi, Nives Melli, and et al. 2021. "Whole Exome Sequencing Is the Minimal Technological Approach in Probands Born to Consanguineous Couples" Genes 12, no. 7: 962. https://doi.org/10.3390/genes12070962