
Wide Fontanels, Delayed Speech Development and Hoarse Voice as Useful Signs in the Diagnosis of KBG Syndrome: A Clinical Description of 23 Cases with Pathogenic Variants Involving the ANKRD11 Gene or Submicroscopic Chromosomal Rearrangements of 16q24.3

 , , , ,
, , , ,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Clinical Investigations
Anthropometric Studies of Children with KBG Syndrome
- (1)
- Head circumference—measured with a tape around the maximum circumference of the head, just above the brow ridges at the front and the most protruding portion of the back of the head (occiput).
- (2)
- Head length (g—op)—from front to back—the distance from the brow ridge (glabella) to the most distant point on the back of the head on the occipital bone (opisthocranion) measured with a spreading caliper.
- (3)
- Head width (eu—eu)—the greatest distance between the most lateral points at the height of the parietal bones (euryon—euryon) measured with a spreading caliper.
- (4)
- Face breadth (zy—zy)—the greatest distance between the even points located most laterally on the zygomatic arches (zygion—zygion) measured with a spreading caliper.
- (5)
- Face height (n—gn)—the linear distance between the depression at the top of the nose (nasion) and the gnathion (on the lower edge of the mandible) measured with a sliding caliper.
- (6)
- Nose length (n—sb)—the distance from the depression at the top of the nose (nasion) to the subnasal point (at the base of the nose) measured with a sliding caliper.
- (7)
- Nose breadth (al—al)—the distance between the most lateral points of the wings of the nose (alare) measured with a sliding caliper.
- (8)
- Interocular breadth (en—en)—the distance between the inner corners of the eyes measured with a sliding caliper.
- (9)
- Biocular breadth (ex—ex)—the distance between the outer corners of the eyes measured with a sliding caliper.
2.2. Cytogenetic and Molecular Investigations
3. Results
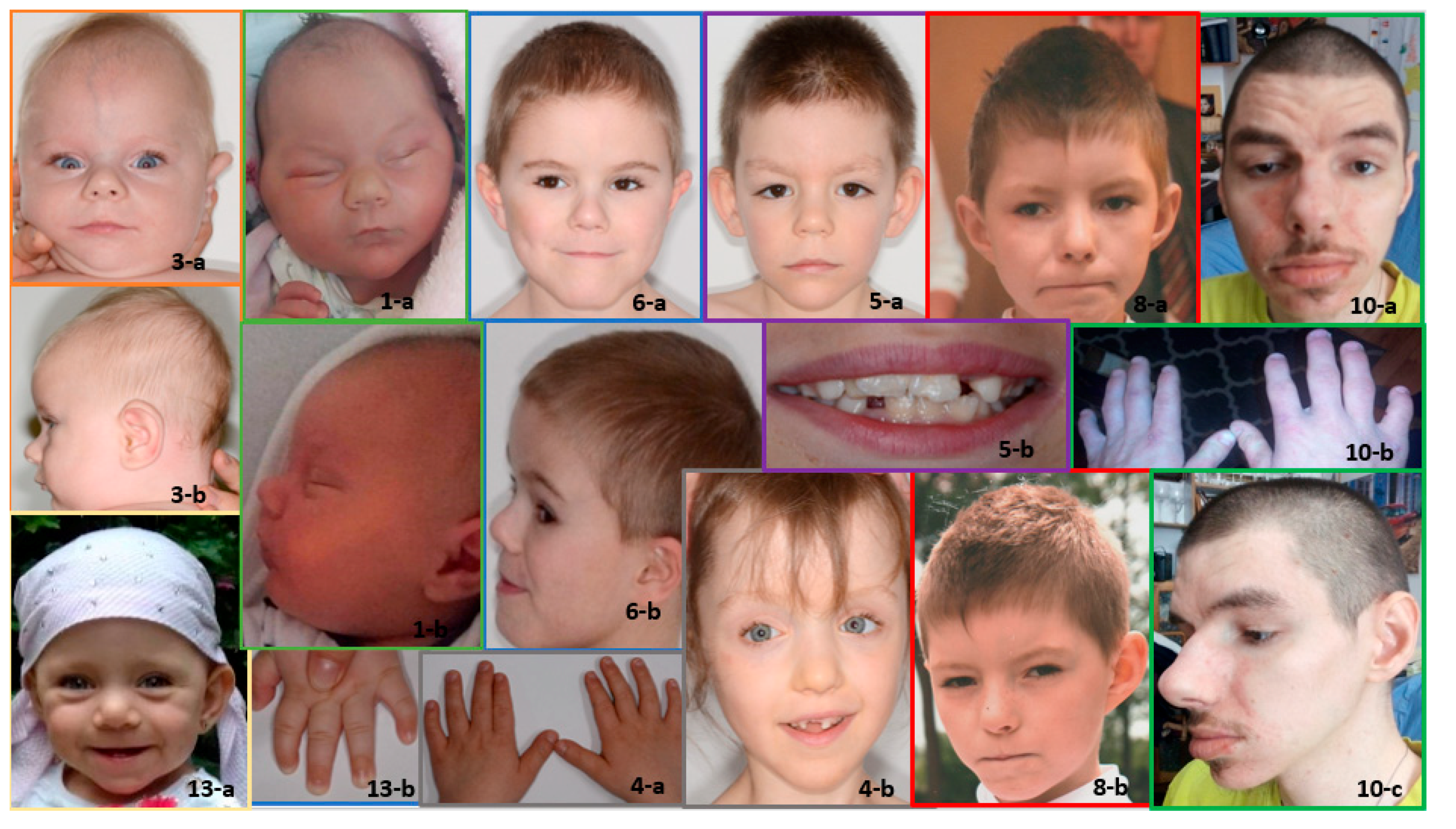
3.1. Clinical Results
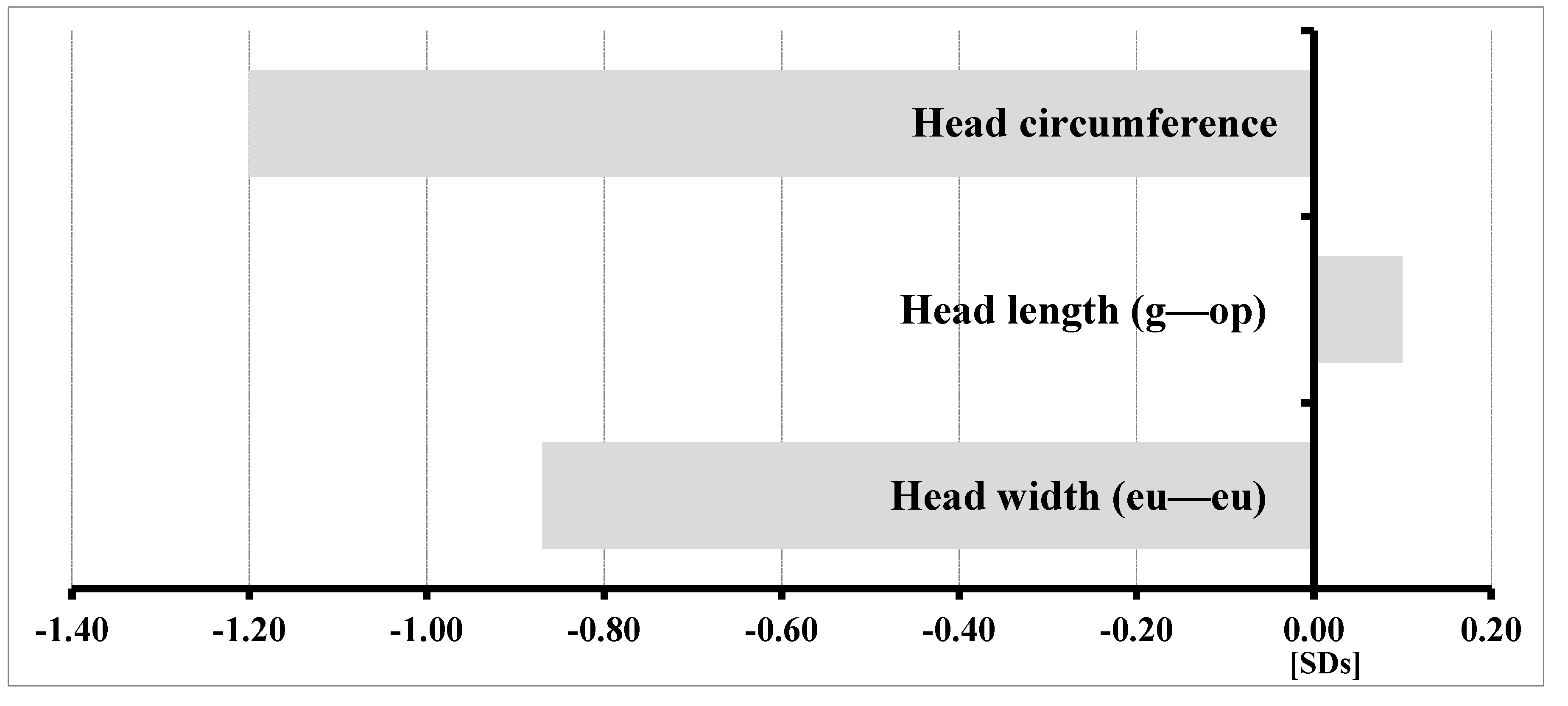
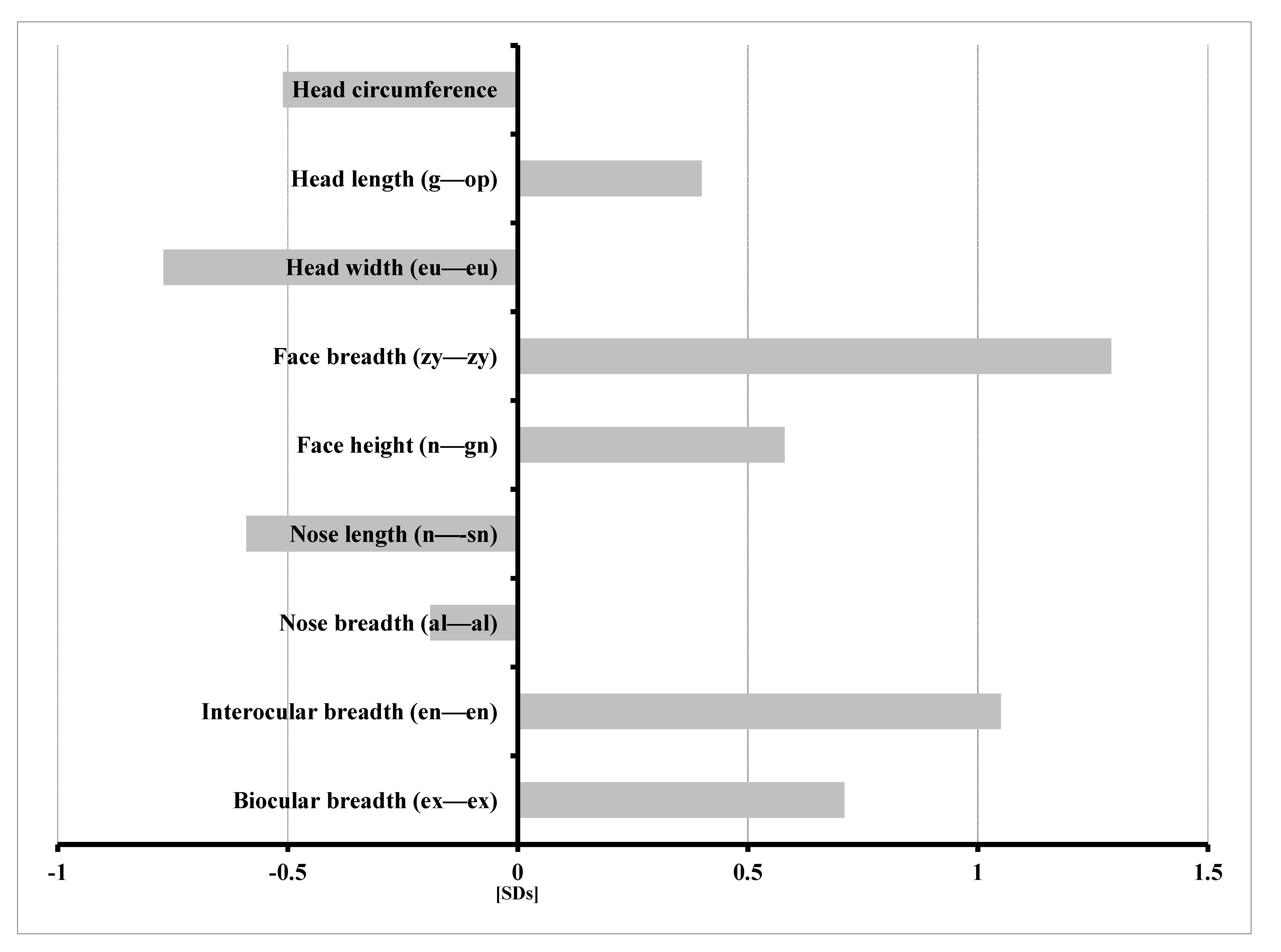
3.2. Results of Anthropometric Studies of Children with KBG Syndrome
- −
- Increased head length (g—op) and facial morphologic length (n—gn);
- −
- Wider face (zy—zy);
- −
- Increased interocular distance—inner canthal (en—en) and outer canthal (ex—ex);
- −
- Decreased head circumference and width (eu—eu); and
- −
- Shorter (n—sn) and narrower (al—al) nose.
3.3. Unexpected Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brancati, F.; D’Avanzo, M.G.; Digilio, M.C.; Sarkozy, A.; Biondi, M.; De Brasi, D.; Mingarelli, R.; Dallapiccola, B. KBG syndrome in a cohort of Italian patients. Am. J. Med. Genet. 2004, 131A, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Brancati, F.; Sarkozy, A.; Dallapiccola, B. KBG syndrome. Orphanet J. Rare Dis. 2006, 1, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Low, K.; Ashraf, T.; Canham, N.; Clayton-Smith, J.; Deshpande, C.; Donaldson, A.; Fisher, R.; Flinter, F.; Foulds, N.; Fryer, A.; et al. Clinical and genetic aspects of KBG syndrome. Am. J. Med. Genet. Part A 2016, 170, 2835–2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldenberg, A.; Riccardi, F.; Tessier, A.; Pfundt, R.; Busa, T.; Cacciagli, P.; Capri, Y.; Coutton, C.; Delahaye-Duriez, A.; Frebourg, T.; et al. Clinical and molecular findings in 39 patients with KBG syndrome caused by deletion or mutation ofANKRD11. Am. J. Med. Genet. Part A 2016, 170, 2847–2859. [Google Scholar] [CrossRef]
- De Bernardi, M.L.; Ivanovski, I.; Caraffi, G.S.; Maini, I.; Street, M.E.; Bayat, A.; Zollino, M.; Lepri, F.R.; Gnazzo, M.; Errichiello, E.; et al. Prominent and elongated coccyx, a new manifestation of KBG syndrome associated with novel mutation in ANKRD11. Am. J. Med. Genet. A 2018, 176, 1991–1995. [Google Scholar] [CrossRef]
- Gnazzo, M.; Lepri, F.R.; Dentici, M.L.; Capolino, R.; Pisaneschi, E.; Agolini, E.; Rinelli, M.; Alesi, V.; Versacci, P.; Genovese, S.; et al. KBG syndrome: Common and uncommon clinical features based on 31 new patients. Am. J. Med. Genet. Part A 2020, 182, 1073–1083. [Google Scholar] [CrossRef]
- Ockeloen, C.W.; Willemsen, M.H.; de Munnik, S.; van Bon, B.W.; de Leeuw, N.; Verrips, A.; Kant, S.G.; Jones, E.A.; Brunner, H.G.; van Loon, R.L.; et al. Further delineation of the KBG syndrome caused by ANKRD11 aberrations. Eur. J. Hum. Genet. 2015, 23, 1270. [Google Scholar] [CrossRef] [Green Version]
- Sirmaci, A.; Spiliopoulos, M.; Brancati, F.; Powell, E.; Duman, D.; Abrams, A.; Bademci, G.; Agolini, E.; Guo, S.; Konuk, B.; et al. Mutations in ANKRD11 Cause KBG Syndrome, Characterized by Intellectual Disability, Skeletal Malformations, and Macrodontia. Am. J. Hum. Genet. 2011, 89, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Youngs, E.L.; Hellings, J.A.; Butler, M.G. ANKRD11 gene deletion in a 17-year-old male. Clin. Dysmorphol. 2011, 20, 170–171. [Google Scholar] [CrossRef] [PubMed]
- Isrie, M.; Hendriks, Y.; Gielissen, N.; Sistermans, E.A.; Willemsen, M.H.; Peeters, H.; Vermeesch, J.R.; Kleefstra, T.; Van Esch, H. Haploinsufficiency of ANKRD11 causes mild cognitive impairment, short stature and minor dysmorphisms. Eur. J. Hum. Genet. 2011, 20, 131–133. [Google Scholar] [CrossRef]
- Khalifa, M.; Stein, J.; Grau, L.; Nelson, V.; Meck, J.; Aradhya, S.; Duby, J. Partial deletion ofANKRD11results in the KBG phenotype distinct from the 16q24.3 microdeletion syndrome. Am. J. Med. Genet. Part A 2013, 161, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Sacharow, S.; Li, D.; Fan, Y.S.; Tekin, M. Familial 16q24.3 microdeletion involving ANKRD11 causes a KBG-like syndrome. Am. J. Med. Genet. Part A 2012, 158A, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, S.; Murakami, A.; Okamoto, N.; Sakamoto, M.; Miyake, N.; Saitsu, H.; Matsumoto, N. A De Novo Deletion at 16q24.3 InvolvingANKRD11in a Japanese Patient with KBG Syndrome. Am. J. Med. Genet. Part A 2013, 161, 1073–1077. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-H.; Seo, E.-J.; Kim, Y.-M.; Cho, H.-J.; Lee, J.-O.; Cheon, C.K.; Yoo, H.-W. A de novo Microdeletion of ANKRD11 Gene in a Korean Patient with KBG Syndrome. Ann. Lab. Med. 2014, 34, 390–394. [Google Scholar] [CrossRef] [Green Version]
- Crippa, M.; Rusconi, D.; Castronovo, C.; Bestetti, I.; Russo, S.; Cereda, A.; Selicorni, A.; Larizza, L.; Finelli, P. Familial intragenic duplication of ANKRD11 underlying three patients of KBG syndrome. Mol. Cytogenet. 2015, 8, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, R.; Saller, K. Lehrbuch der Anthropologie; Gustav Fisher Verlag: Stuttgart, Germany, 1957. [Google Scholar]
- Malinowski, A.; Bożiłow, W. Podstawy Antropometrii: Metody, Techniki, Normy; Wydawnictwo Naukowe PWN: Warszawa-Łódź, Poland, 1997. [Google Scholar]
- Niedźwiecka, Z.; Palczewska, I. Wskaźniki Rozwoju Somatycznego Dzieci i Młodzieży Warszawskiej. In Medycyna Wieku Rozwojowego; Instytut Matki i Dziecka: Warszawa, Poland, 2001; Volume 5, Suplement I. [Google Scholar]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F.; Gu, B.; Hart, J.; et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Liu, X.; Li, C.; Mou, C.; Dong, Y.; Tu, Y. dbNSFP v4: A comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 2020, 12, 1–8. [Google Scholar] [CrossRef]
- Mattei, D.; Cavarzere, P.; Gaudino, R.; Antoniazzi, F.; Piacentini, G. DYSMORPHIC features and adult short stature: Possible clinical markers of KBG syndrome. Ital. J. Pediatr. 2021, 47, 1–6. [Google Scholar] [CrossRef]
- Friedman, J.; Hanson, J.W.; Graham, C.B.; Smith, D.W. Saethre-Chotzen syndrome: A broad and variable pattern of skeletal malformations. J. Pediatr. 1977, 91, 929–933. [Google Scholar] [CrossRef]
- Spruijt, B.; Rijken, B.F.M.; Joosten, K.F.M.; Bredero-Boelhouwer, H.H.; Pullens, B.; Lequin, M.H.; Wolvius, E.B.; Van Veelen-Vincent, M.L.C.; Mathijssen, I.M.J. Atypical presentation of a newborn with Apert syndrome. Child’s Nerv. Syst. 2015, 31, 481–486. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | KBG Gene Mutation/16q24 Rearrangement | Pathogenicity | Age of Diagnosis (Years) | Poor Weight Gain/Feeding Problems | Short Stature | Macrodontia | Hoarse Voice | Behavioral Problems | DD/ID | Delayed Speech | Wide Fontanel, Delayed Closure |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | c.1903_1907del (p.Lys635Glnfs*26) | Pathogenic (HGMD: CD150411) | 0.3 | − | + (−2.14 SDs) | NA | + | NA | − | NA | + |
| 2 | c.1903_1907del (p.Lys635Glnfs*26) | Pathogenic (HGMD: CD150411) | 2.6 | − | − (−0.52 SDs) | + | + | − | − | + | + |
| 3 | c.7607G>A (p.Arg2538Gln) | Pathogenic (de novo) | 0.3 | + | − (−1.66 SDs) | NA | − | NA | − | NA | + |
| 4 | c.4558del (p.Asp1520Thrfs*11) | Pathogenic (de novo) | 7 | + | + (−3.61 SDs) | + | + | + | − | + 3 words | + |
| 5 | c.2395A>T (p.Lys799Ter) | Pathogenic (de novo) | 4.4 | + | − (−0.48 SDs) | − | + | + | Delayed motor development | + | + |
| 6 | c.1389dup (p.Gly464Argfs*29) | Pathogenic (de novo) | 4 | + | + | − | + | + | + | + | + |
| 7 | c. 7552C>T (p.Gln2518Ter) | Pathogenic (ClinVar: 280739) | 10 | − | + (−2.5 SDs) | + | + | + | Learning problems | + | + |
| 8 | c.2828_2829del (p.Glu943Valfs*74) | Pathogenic | 16 | − | − (−1.59 SDs) | + | + | + | + | + | + |
| 9 | c.6340C>T (p.Gln2114Ter) | Pathogenic (de novo) | 8 | + | − | + | − | + | + | + | − |
| 10 | c.3295_3296del (p.Phe1099Leufs*2) | Pathogenic (de novo) | 26 | + | − | + | + | − | + | + | - |
| 11 | c.3771dup (p.Glu1258Argfs*25) | Pathogenic | 8 | + | + | ND | − | + | + mild | + | − |
| 12 | c.1385_1388del (p.Thr462Lysfs*47) | Pathogenic (HGMD: CD1412989) | 22 | + | − | + | + | + | + moderate ID | + | + |
| 13 | c.6053_6057del (p.Pro2018Argfs*12) | Pathogenic | 3 | + | polydactyly unilateral | + | − | + | DD epilepsy | + | + |
| 14 | arr[hg38]13q21.31(61968361-63533241)x1; 16q24.2q24.3(87921245-89417758)x1 | Pathogenic | 8 | + | − (−1.02 SDs) | + | − | + | Learning problems | + | − |
| 15 | arr[hg38] 16q24.3(89171712-89274753)x1 | Pathogenic (de novo) | 5.5 | − | − (−1.71 SDs) | − | + | − | IQ-nl | − | + |
| 16 | arr[hg38]16q24.3(89277485-89517986)x1 | Unknown pathogenicity | 3.5 | − | − | − | − | + | DD | + | + |
| 17 | arr[hg38] 16q24.3(89195406-89489612)x3 | Pathogenic | 6 | − | − | − | − | + ASD | − | + | − |
| 18 | arr[hg38]16q24.3(89266045-89305443)x1 | mosaic | 5 | + | − (−1.08 SDs) | − | + | + | − | − | − |
| 19 | 16q24.3(89277485-89431539)x1 | Pathogenic(de novo) | 6 | + | − | − | − | + | Mild ID | + | + |
| 20 | arr[hg38] 16q24.3(89458995-89487166)x1 | Unknown pathogenicity | 22 | − | + (−3.3 SDs) | − | − | + | Learning problems | − | ND |
| 21 | arr[hg38] 16q24.3(89481147-89489612)x1 | Unknown pathogenicity | 15 | + | + (−3.37 SDs) | + | − | + | Learning problems | + | − |
| 22 | arr[hg38] 16q24.3(89409759-89418313)x1 | Likely pathogenic/Pathogenic | 5 | − | − | − | − | ASD | DD | + | Partial sagittal craniosynostosis |
| 23 | arr[hg38] 16q24.3(89277485-89489140))x1 | Pathogenic | 13 | + | + | + | + | ADHD | Mild ID | + | − |
| Patient | Age | Weight | Height (Length) | BMI | Head Circumference | Chest Circumference | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Years | kg | Z-Score | cm | Z-Score | kg/m2 | Z-Score | cm | Z-Score | cm | Z-Score | |
| 1 | 0.3 | 5.10 | −1.75 | 58.6 | −2.14 | 14.85 | −1.04 | 38.9 | −1.82 | ||
| 3 | 1.4 | 11.3 | −0.09 | 79.4 | −0.77 | 17.80 | 0.58 | 45.7 | −2.04 | 49.8 | 0.72 |
| 2 | 2.6 | 10.4 | −2.50 | 91.0 | −0.52 | 12.56 | −3.13 | 47.1 | −2.36 | 47.0 | −1.76 |
| 5 | 4.4 | 16.4 | −0.88 | 106.0 | −0.48 | 14.60 | −0.79 | 50.0 | −1.27 | 51.6 | −0.98 |
| 9 | 5.4 | 17.1 | −0.93 | 111.0 | −0.48 | 14.12 | −0.89 | 48.6 | −1.73 | 50.5 | −1.23 |
| 6 | 5.9 | 18.5 | −0.93 | 110.0 | −1.48 | 15.29 | −0.31 | 50.0 | −1.58 | 55.0 | −0.48 |
| 11 | 6.8 | 17.7 | −1.89 | 112.5 | −2.19 | 13.99 | −1.15 | 48.4 | −3.05 | 54.7 | −1.30 |
| 15 | 7.2 | 21.9 | −0.59 | 114.5 | −1.51 | 16.73 | 0.24 | 50.5 | −0.73 | 56.2 | −0.37 |
| 17 | 7.3 | 26.8 | 0.16 | 125.1 | −0.28 | 17.12 | 0.46 | 53.6 | 0.57 | 59.5 | −0.05 |
| 7 | 11.3 | 40.8 | −0.12 | 141.8 | −1.13 | 20.29 | 0.59 | 56.7 | 1.48 | 73.2 | 0.50 |
| 8 | 14.8 | 53.4 | −0.73 | 160.5 | −1.59 | 20.34 | 0.00 | 55.0 | −0.67 | 79.6 | −0.08 |
| Mean z-score | −0.93 | −1.14 | −0.49 | −1.20 | −0.50 | ||||||
| Clinical Signs | Syndrome/Gene |
|---|---|
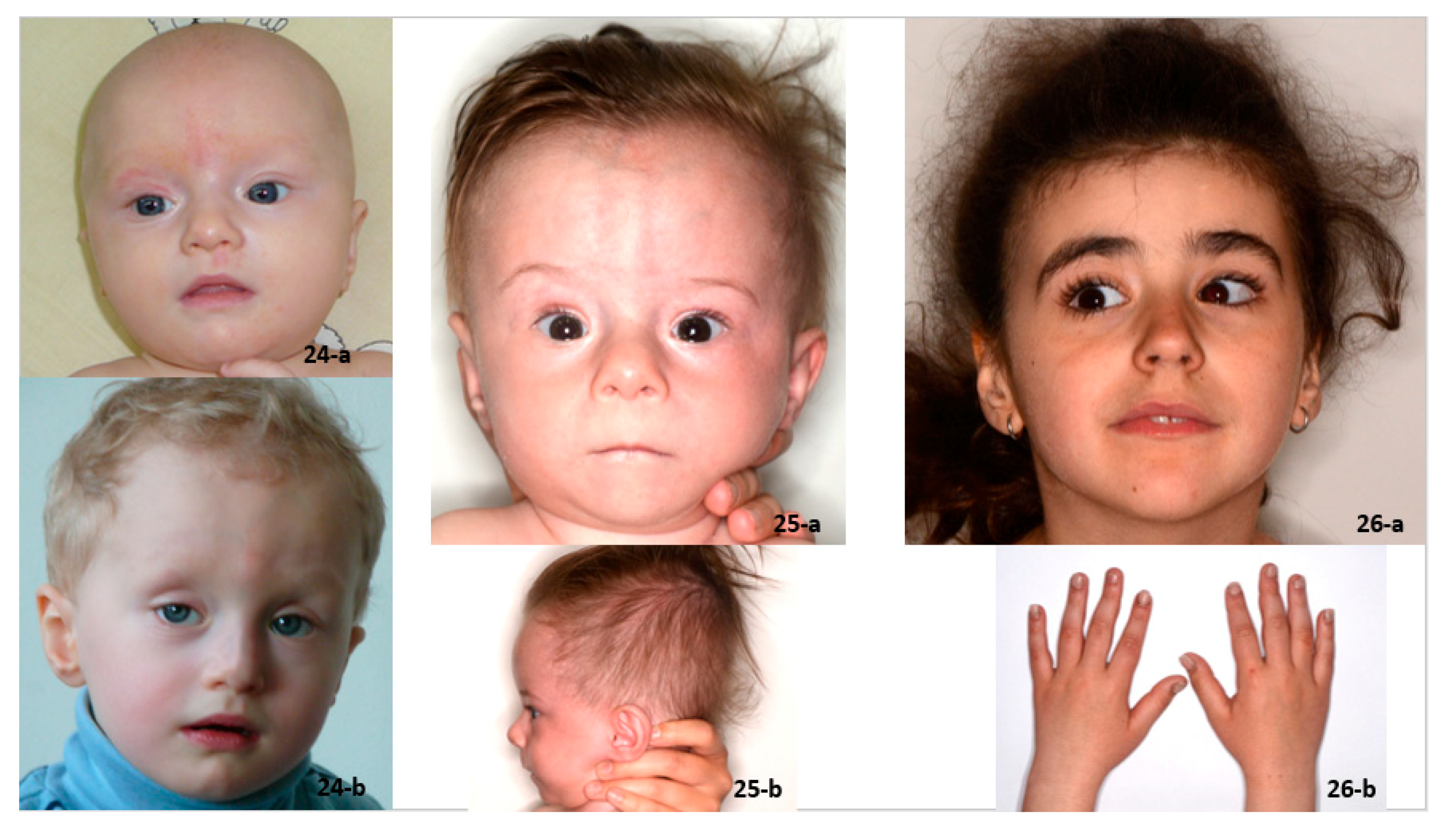
| Patient 24: Intellectual disability, dysphonic voice, dysmorphic features | Weiss–Kruszka syndrome ZNF462: NM_021224.6:c.4784_4785del; p.(His1595Leufs*9) |
| Patient 25: IUGR, short stature (−3.83 SDs), developmental and speech delay, dysmorphic features | Keipert syndrome GPC4: NM_001448.3:c.881C>T; p.(Ala294Val) |
| Patient 26: short normal stature (−1.83 SDs), hoarse voice, hirsutism, psychomotor hyperactivity. | Pierpont syndrome TBL1XR1: NM_024665.7: c.513A>C; p.(Glu171Asp) |
| Unexpected results of aCGH: | |
| 2 patients with 16q24.3 deletion (Patient 20 and Patient 21) | |
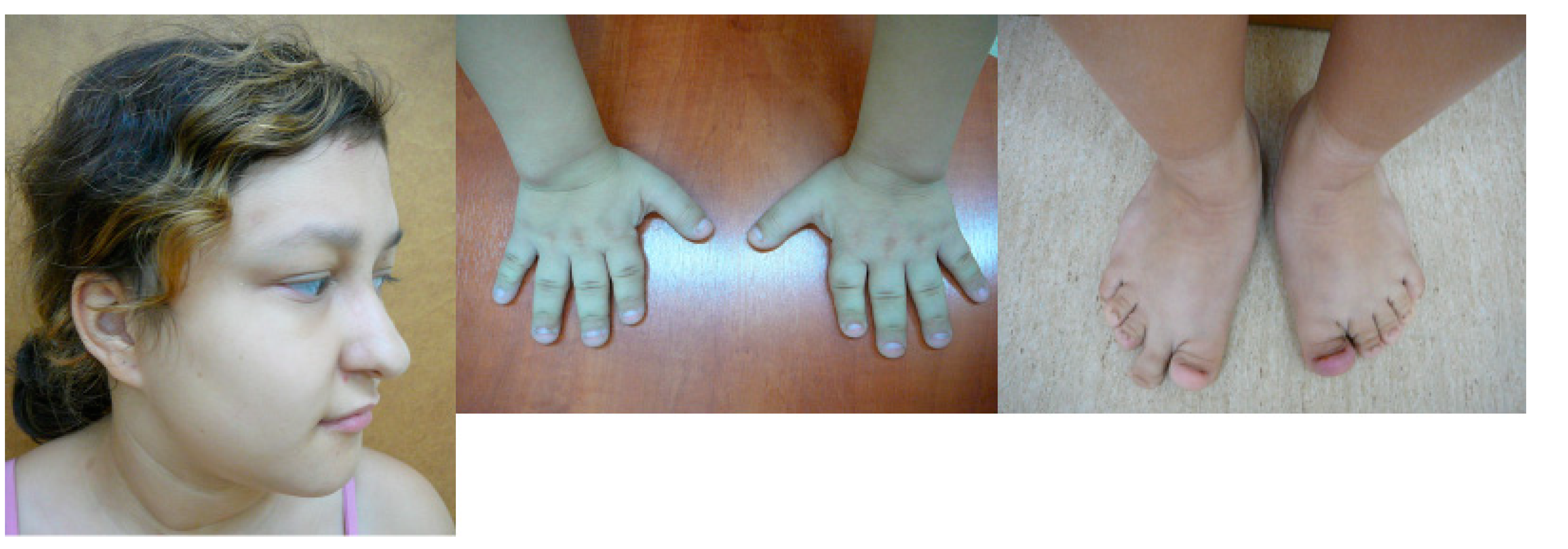
| Short stature below −3.3 SDs Similar dysmorphic features (Figure 5): long nose, long face Learning problems, psychomotor hyperactivity Macrodontia in one patient Severe brachydactyly in one patient with a variant of unknown significance in the TRPS1 gene: NM_014112.4:c.2834C>T (p.(Pro945Leu) | Both patients with microdeletions of unknown pathogenicity localized in intron 1 of the ANKRD11 gene Analyses of parents’ DNA not performed |
| Patient 17 with de novo 16q24.3 duplication | |
| Autism spectrum disorder, stereotypic movements, speech delay | Duplication of: SLC22A31, ZNF778 and ANKRD11 of unknown pathogenicity |
| Most Common |
| Achondroplasia |
| Congenital hypothyroidism |
| Down Syndrome |
| Less Common |
| Skeletal Disorders |
| Cleidoclanial dysplasia |
| Acrocallosal syndrome |
| Campomelic dysplasia |
| Hypophosphatasia |
| Kenny-Caffey syndrome |
| Osteogenesis imperfecta |
| Dysostosis Stanescu type |
| Dysmorphogenetic Syndromes |
| KBG syndrome |
| Robinow syndrome |
| Beckwith-Wiedemann syndrome |
| Zellweger syndrome |
| Cutis laxa |
| VATER association |
| Otopalatodigital syndrome |
| Occipital horn syndrome |
| Autosomal recessive cutis laxa type 2A |
| Partial trisomy of the short arm of chromosome 9 |
| KBG syndrome—low pitched hoarse voice |
| Williams Syndrome—low pitched hoarse voice |
| Smith–Magenis syndrome—low pitched hoarse deep voice |
| Congenital Hypothyroidism—low pitched hoarse deep voice |
| Genetic metabolic diseases—mucopolysaccharidosis, Farber diseases, disseminated lipogranulomatosis, lipoid protenois, Morquio A |
| Cutis laxa (pendulous skin and hoarse cry)—vocal folds thickening |
| Dubowitz syndrome—high pitched hoarse voice |
| Idiopathic familial voice disorder—vocal fold paralysis |
| Ehlers–Danlos syndrome type VIII—hoarse voice |
| Costello syndrome—hoarse voice |
| Aicardi–Goutieres syndrome—low pitched hoarse voice |
| Werner syndrome—hoarse voice |
| X-linked hypohidrotic ectodermal dysplasia (XLHED)—hoarse raspy voice |
| Major Criteria | Proportion of Patients in this Cohort with Feature | Children below 7 Years of Age | Children below 2 Year of Age |
|---|---|---|---|
| Psychomotor hyperactivity/ADHD | 85.7% | 90% | - |
| Speech delay | 80.9% | 82% | - |
| Gain weight problems with good appetite | 56% | 80% | - |
| Height below the 10th centile | 56% | 40% | 25% |
| Wide, delayed closing anterior fontanel | 56% | 69% | 100% |
| Hoarse voice | 56% | 69% | 75% |
| Macrodontia | 52% | - | - |
| 1st degree relative with KBG Syndrome | 0.0% | no relatives available | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kutkowska-Kaźmierczak, A.; Boczar, M.; Kalka, E.; Castañeda, J.; Klapecki, J.; Pietrzyk, A.; Barczyk, A.; Malinowska, O.; Landowska, A.; Gambin, T.; et al. Wide Fontanels, Delayed Speech Development and Hoarse Voice as Useful Signs in the Diagnosis of KBG Syndrome: A Clinical Description of 23 Cases with Pathogenic Variants Involving the ANKRD11 Gene or Submicroscopic Chromosomal Rearrangements of 16q24.3. Genes 2021, 12, 1257. https://doi.org/10.3390/genes12081257
Kutkowska-Kaźmierczak A, Boczar M, Kalka E, Castañeda J, Klapecki J, Pietrzyk A, Barczyk A, Malinowska O, Landowska A, Gambin T, et al. Wide Fontanels, Delayed Speech Development and Hoarse Voice as Useful Signs in the Diagnosis of KBG Syndrome: A Clinical Description of 23 Cases with Pathogenic Variants Involving the ANKRD11 Gene or Submicroscopic Chromosomal Rearrangements of 16q24.3. Genes. 2021; 12(8):1257. https://doi.org/10.3390/genes12081257
Chicago/Turabian StyleKutkowska-Kaźmierczak, Anna, Maria Boczar, Ewa Kalka, Jennifer Castañeda, Jakub Klapecki, Aleksandra Pietrzyk, Artur Barczyk, Olga Malinowska, Aleksandra Landowska, Tomasz Gambin, and et al. 2021. "Wide Fontanels, Delayed Speech Development and Hoarse Voice as Useful Signs in the Diagnosis of KBG Syndrome: A Clinical Description of 23 Cases with Pathogenic Variants Involving the ANKRD11 Gene or Submicroscopic Chromosomal Rearrangements of 16q24.3" Genes 12, no. 8: 1257. https://doi.org/10.3390/genes12081257