
Agenesis of the Corpus Callosum with Facial Dysmorphism and Intellectual Disability in Sibs Associated with Compound Heterozygous KDM5B Variants

 , , and
, , and 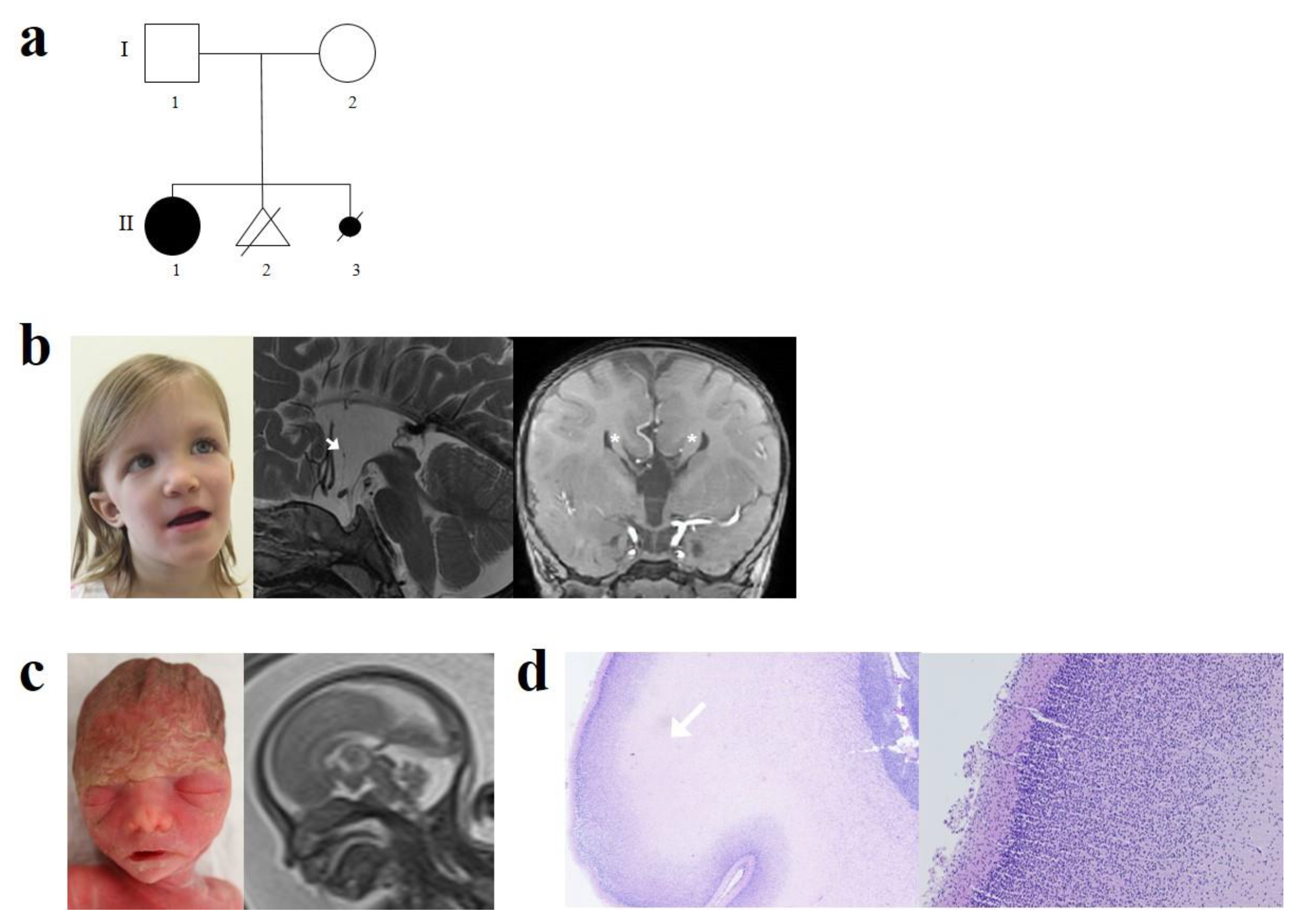{kind=link}
Abstract
:1. Introduction
2. Case Reports
3. Genetic Studies
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Edwards, T.; Sherr, E.H.; Barkovich, A.J.; Richards, L.J. Clinical, genetic and imaging findings identify new causes for corpus callosum development syndromes. Brain 2014, 137, 1579–1613. [Google Scholar] [CrossRef] [Green Version]
- D’Antonio, F.; Pagani, G.; Familiari, A.; Khalil, A.; Sagies, T.-L.; Malinger, G.; Leibovitz, Z.; Garel, C.; Moutard, M.L.; Pilu, G.; et al. Outcomes Associated With Isolated Agenesis of the Corpus Callosum: A Meta-analysis. Pediatrics 2016, 138, e20160445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, E.E.; Mowat, D. Agenesis of the corpus callosum: A clinical approach to diagnosis. Am. J. Med Genet. Part C Semin. Med. Genet. 2014, 166, 184–197. [Google Scholar] [CrossRef] [PubMed]
- De Wit, M.C.; Boekhorst, F.; Mancini, G.M.; Smit, L.S.; Groenenberg, I.A.L.; Dudink, J.; de Vries, F.A.T.; Go, A.T.J.I.; Galjaard, R.J.H. Advanced genomic testing may aid in counseling of isolated agenesis of the corpus callosum on prenatal ultrasound. Prenat. Diagn. 2017, 37, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Al-Mubarak, B.; Abouelhoda, M.; Omar, A.; Aldhalaan, H.; Aldosari, M.; Nester, M.; Alshamrani, H.A.; El-Kalioby, M.; Goljan, E.; Albar, R.; et al. Whole exome sequencing reveals inherited and de novo variants in autism spectrum disorder: A trio study from Saudi families. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Athanasakis, E.; Licastro, D.; Faletra, F.; Fabretto, A.; Dipresa, S.; Vozzi, D.; Morgan, A.; D’Adamo, A.P.; Pecile, V.; Biarnés, X.; et al. Next generation sequencing in nonsyndromic intellectual disability: From a negative molecular karyotype to a possible causative mutation detection. Am. J. Med Genet. Part A 2014, 164, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Faundes, V.; Newman, W.; Bernardini, L.; Canham, N.; Clayton-Smith, J.; Dallapiccola, B.; Davies, S.J.; Demos, M.K.; Goldman, A.; Gill, H.; et al. Histone Lysine Methylases and Demethylases in the Landscape of Human Developmental Disorders. Am. J. Hum. Genet. 2018, 102, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.; Sanders, S.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebrun, N.; Mehler-Jacob, C.; Poirier, K.; Zordan, C.; Lacombe, D.; Carion, N.; Billuart, P.; Bienvenu, T. Novel KDM5B splice variants identified in patients with developmental disorders: Functional consequences. Gene 2018, 679, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.C.; Jones, W.D.; McIntyre, R.; Sanchez-Andrade, G.; Sanderson, M.; Stephenson, J.D.; Jones, C.P.; Handsaker, J.; Gallone, G.; Bruntraeger, M.; et al. Quantifying the contribution of recessive coding variation to developmental disorders. Science 2018, 362, 1161–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miolo, G.; Giuffrida, M.G.; Corona, G.; Capalbo, A.; Pivetta, B.; Tessitori, G.; Bernardini, L. A novel mosaic 1q32.1 microduplication identified through Chromosome Microarray Analysis: Narrowing the smallest critical region including KDM5B gene found associated with neurodevelopmetal disorders. Eur. J. Med. Genet. 2018, 62, 103558. [Google Scholar] [CrossRef] [PubMed]
- Hetts, S.W.; Sherr, E.H.; Chao, S.; Gobuty, S.; Barkovich, A.J. Anomalies of the Corpus Callosum: An MR Analysis of the Phenotypic Spectrum of Associated Malformations. Am. J. Roentgenol. 2006, 187, 1343–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesaretti, C.; Nanni, M.; Ghi, T.; Parazzini, C.; Conte, G.; Contro, E.; Grisolia, G.; Righini, A. Variability of Forebrain Commissures in Callosal Agenesis: A Prenatal MR Imaging Study. Am. J. Neuroradiol. 2015, 37, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehead, M.; Najim, N. Thalamic Massa Intermedia in Children with and without Midline Brain Malformations. Am. J. Neuroradiol. 2020, 41, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Vallianatos, C.N.; Iwase, S. Disrupted intricacy of histone H3K4 methylation in neurodevelopmental disorders. Epigenomics 2015, 7, 503–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Obana, E.A.; Radomski, K.L.; Sukumar, G.; Wynder, C.; Dalgard, C.L.; Doughty, M.L. Inhibition of the histone demethylase Kdm5b promotes neurogenesis and derepresses Reln (reelin) in neural stem cells from the adult subventricular zone of mice. Mol. Biol. Cell 2016, 27, 627–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fame, R.; MacDonald, J.; Macklis, J.D. Development, specification, and diversity of callosal projection neurons. Trends Neurosci. 2011, 34, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcamo, E.A.; Chirivella, L.; Dautzenberg, M.; Dobreva, G.; Fariñas, I.; Grosschedl, R.; McConnell, S.K. Satb2 Regulates Callosal Projection Neuron Identity in the Developing Cerebral Cortex. Neuron 2008, 57, 364–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebon, S.; Quinodoz, M.; Peter, V.G.; Gengler, C.; Blanchard, G.; Cina, V.; Campos-Xavier, B.; Rivolta, C.; Superti-Furga, A. Agenesis of the Corpus Callosum with Facial Dysmorphism and Intellectual Disability in Sibs Associated with Compound Heterozygous KDM5B Variants. Genes 2021, 12, 1397. https://doi.org/10.3390/genes12091397
Lebon S, Quinodoz M, Peter VG, Gengler C, Blanchard G, Cina V, Campos-Xavier B, Rivolta C, Superti-Furga A. Agenesis of the Corpus Callosum with Facial Dysmorphism and Intellectual Disability in Sibs Associated with Compound Heterozygous KDM5B Variants. Genes. 2021; 12(9):1397. https://doi.org/10.3390/genes12091397
Chicago/Turabian StyleLebon, Sébastien, Mathieu Quinodoz, Virginie G. Peter, Carole Gengler, Gaëlle Blanchard, Viviane Cina, Belinda Campos-Xavier, Carlo Rivolta, and Andrea Superti-Furga. 2021. "Agenesis of the Corpus Callosum with Facial Dysmorphism and Intellectual Disability in Sibs Associated with Compound Heterozygous KDM5B Variants" Genes 12, no. 9: 1397. https://doi.org/10.3390/genes12091397