
Whole-Exome Sequencing and Copy Number Analysis in a Patient with Warburg Micro Syndrome
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Clinical Assessments
2.3. Exome Sequencing and Bioinformatic Analysis
2.4. Pathogenicity Assessments of the Variants
3. Results
3.1. Clinical Phenotypes
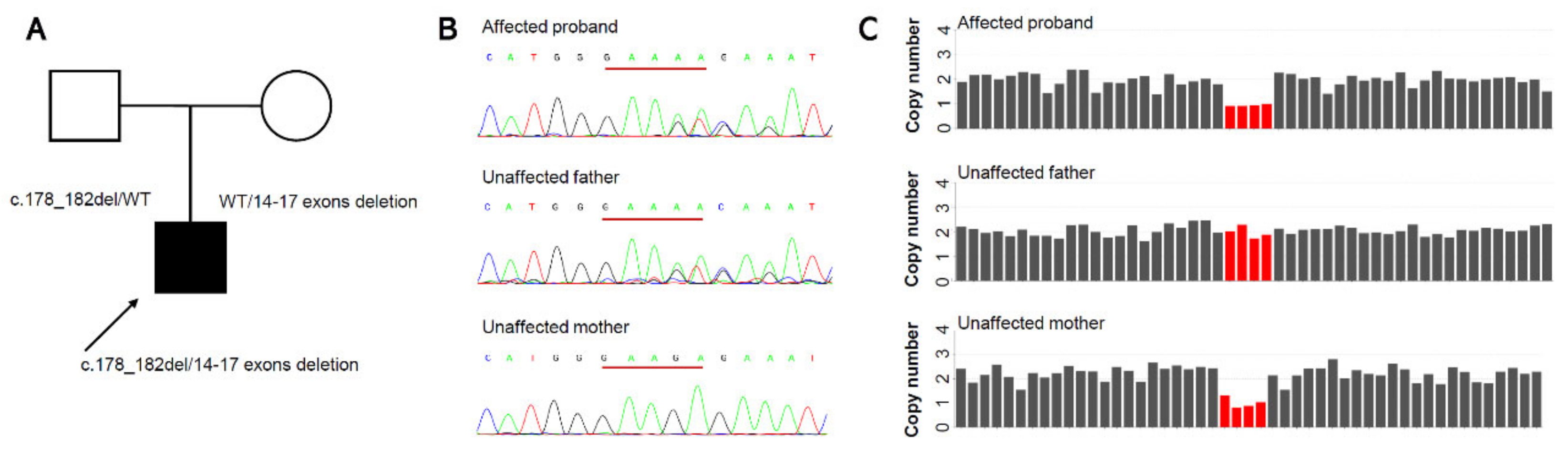
3.2. Molecular Findings
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aligianis, I.A.; Johnson, C.A.; Gissen, P.; Chen, D.; Hampshire, D.; Hoffmann, K.; Maina, E.N.; Morgan, N.V.; Tee, L.; Morton, J.; et al. Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nat. Genet. 2005, 37, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hamid, M.S.; Abdel-Ghafar, S.F.; Ismail, S.R.; Desouky, L.M.; Issa, M.Y.; Effat, L.K.; Zaki, M.S. Micro and Martsolf syndromes in 34 new patients: Refining the phenotypic spectrum and further molecular insights. Clin. Genet. 2020, 98, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Bem, D.; Yoshimura, S.; Nunes-Bastos, R.; Bond, F.C.; Kurian, M.A.; Rahman, F.; Handley, M.T.; Hadzhiev, Y.; Masood, I.; Straatman-Iwanowska, A.A.; et al. Loss-of-function mutations in RAB18 cause Warburg micro syndrome. Am. J. Hum. Genet. 2011, 88, 499–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liegel, R.P.; Handley, M.T.; Ronchetti, A.; Brown, S.; Langemeyer, L.; Linford, A.; Chang, B.; Morris-Rosendahl, D.J.; Carpanini, S.; Posmyk, R.; et al. Loss-of-function mutations in TBC1D20 cause cataracts and male infertility in blind sterile mice and Warburg micro syndrome in humans. Am. J. Hum. Genet. 2013, 93, 1001–1014. [Google Scholar] [CrossRef] [Green Version]
- Aligianis, I.A.; Morgan, N.V.; Mione, M.; Johnson, C.A.; Rosser, E.; Hennekam, R.C.; Adams, G.; Trembath, R.C.; Pilz, D.T.; Stoodley, N.; et al. Mutation in Rab3 GTPase-activating protein (RAB3GAP) noncatalytic subunit in a kindred with Martsolf syndrome. Am. J. Hum. Genet. 2006, 78, 702–707. [Google Scholar] [CrossRef] [Green Version]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Handley, M.T.; Carpanini, S.M.; Mali, G.R.; Sidjanin, D.J.; Aligianis, I.A.; Jackson, I.J.; FitzPatrick, D.R. Warburg Micro syndrome is caused by RAB18 deficiency or dysregulation. Open. Biol. 2015, 5, 150047. [Google Scholar] [CrossRef]
- Dejgaard, S.Y.; Presley, J.F. Rab18: New insights into the function of an essential protein. Cell. Mol. Life Sci. 2019, 76, 1935–1945. [Google Scholar] [CrossRef]
- Lin, H.; Long, E.; Chen, W.; Liu, Y. Documenting rare disease data in China. Science 2015, 349, 1064. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, X.; Qin, T.; Wang, D.; Lin, X.; Zhu, Y.; Tan, H.; Zhao, L.; Li, J.; Lin, Z.; et al. Unusual Presentation in WAGR Syndrome: Expanding the Phenotypic and Genotypic Spectrum of the Diseases. Gene 2022, 13, 1431. [Google Scholar] [CrossRef]
- Wang, Q.; Qin, T.; Tan, H.; Ding, X.; Lin, X.; Li, J.; Lin, Z.; Sun, L.; Lin, H.; Chen, W. Broadening the genotypic and phenotypic spectrum of MAF in three Chinese Han congenital cataracts families. Am. J. Med. Genet. A 2022, 188, 2888–2898. [Google Scholar] [CrossRef] [PubMed]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; He, N.; Zhang, J.W.; Lin, Z.J.; Wang, J.; Yan, L.M.; Meng, H.; Tang, B.; Li, B.M.; Liu, X.R.; et al. Novel mutations and phenotypes of epilepsy-associated genes in epileptic encephalopathies. Genes Brain Behav. 2018, 17, e12456. [Google Scholar] [CrossRef]
- Tavtigian, S.V.; Greenblatt, M.S.; Harrison, S.M.; Nussbaum, R.L.; Prabhu, S.A.; Boucher, K.M.; Biesecker, L.G.; ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet. Med. 2018, 20, 1054–1060. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- WHO. WHO Child Growth Standards: Length/Height-for-Age, Weight-for-Age, Weight-for-Length, Weight-for-Height and Body Mass Index-for-Age; World Health Organization (WHO): Geneva, Switzerland, 2006. [Google Scholar]
- Brandt, T.; Sack, L.M.; Arjona, D.; Tan, D.; Mei, H.; Cui, H.; Gao, H.; Bean, L.J.H.; Ankala, A.; Del Gaudio, D.; et al. Adapting ACMG/AMP sequence variant classification guidelines for single-gene copy number variants. Genet. Med. 2020, 22, 336–344. [Google Scholar] [CrossRef]
- Handley, M.T.; Morris-Rosendahl, D.J.; Brown, S.; Macdonald, F.; Hardy, C.; Bem, D.; Carpanini, S.M.; Borck, G.; Martorell, L.; Izzi, C.; et al. Mutation spectrum in RAB3GAP1, RAB3GAP2, and RAB18 and genotype-phenotype correlations in warburg micro syndrome and Martsolf syndrome. Hum. Mutat. 2013, 34, 686–696. [Google Scholar] [CrossRef]
- Koparir, A.; Karatas, O.F.; Yilmaz, S.S.; Suer, I.; Ozer, B.; Yuceturk, B.; Ozen, M. Revealing the functions of novel mutations in RAB3GAP1 in Martsolf and Warburg micro syndromes. Am. J. Med. Genet. A 2019, 179, 579–587. [Google Scholar] [CrossRef]
- Hozhabri, H.; Talebi, M.; Mehrjardi, M.Y.V.; De Luca, A.; Dehghani, M. Martsolf syndrome with novel mutation in the TBC1D20 gene in a family from Iran. Am. J. Med. Genet. A 2020, 182, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, P.K.; Premalatha, R.; Sabapathy, S. Warburg micro syndrome in siblings from India. J. Pediatr. Neurosci. 2016, 11, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Kerkeni, N.; Kharrat, M.; Maazoul, F.; Boudabous, H.; M’Rad, R.; Trabelsi, M. Novel RAB3GAP1 Mutation in the First Tunisian Family With Warburg Micro Syndrome. J. Clin. Neurol. 2022, 18, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Mutlu Albayrak, H.; Elcioglu, N.H.; Yeter, B.; Karaer, K. From cataract to syndrome diagnosis: Revaluation of Warburg-Micro syndrome Type 1 patients. Am. J. Med. Genet. A 2021, 185, 2325–2334. [Google Scholar] [CrossRef] [PubMed]
- Haargaard, B.; Wohlfahrt, J.; Fledelius, H.C.; Rosenberg, T.; Melbye, M. Incidence and cumulative risk of childhood cataract in a cohort of 2.6 million Danish children. Invest. Ophthalmol. Vis. Sci. 2004, 45, 1316–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiels, A.; Bennett, T.M.; Hejtmancik, J.F. Cat-Map: Putting cataract on the map. Mol. Vis. 2010, 16, 2007–2015. [Google Scholar] [PubMed]
- Gillespie, R.L.; O’Sullivan, J.; Ashworth, J.; Bhaskar, S.; Williams, S.; Biswas, S.; Kehdi, E.; Ramsden, S.C.; Clayton-Smith, J.; Black, G.C.; et al. Personalized diagnosis and management of congenital cataract by next-generation sequencing. Ophthalmology 2014, 121, 2124–2137. [Google Scholar] [CrossRef]
- Rechsteiner, D.; Issler, L.; Koller, S.; Lang, E.; Bahr, L.; Feil, S.; Ruegger, C.M.; Kottke, R.; Toelle, S.P.; Zweifel, N.; et al. Genetic Analysis in a Swiss Cohort of Bilateral Congenital Cataract. JAMA Ophthalmol. 2021, 139, 691–700. [Google Scholar] [CrossRef]
- Bell, S.; Malka, S.; Lloyd, I.C.; Moosajee, M. Clinical Spectrum and Genetic Diagnosis of 54 Consecutive Patients Aged 0-25 with Bilateral Cataracts. Genes 2021, 12, 131. [Google Scholar] [CrossRef]
- Sun, W.; Xiao, X.; Li, S.; Guo, X.; Zhang, Q. Exome sequencing of 18 Chinese families with congenital cataracts: A new sight of the NHS gene. PLoS ONE 2014, 9, e100455. [Google Scholar] [CrossRef]
- Kandaswamy, D.K.; Prakash, M.V.S.; Graw, J.; Koller, S.; Magyar, I.; Tiwari, A.; Berger, W.; Santhiya, S.T. Application of WES Towards Molecular Investigation of Congenital Cataracts: Identification of Novel Alleles and Genes in a Hospital-Based Cohort of South India. Int. J. Mol. Sci. 2020, 21, 9569. [Google Scholar] [CrossRef] [PubMed]
- Boone, P.M.; Bacino, C.A.; Shaw, C.A.; Eng, P.A.; Hixson, P.M.; Pursley, A.N.; Kang, S.H.; Yang, Y.; Wiszniewska, J.; Nowakowska, B.A.; et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum. Mutat. 2010, 31, 1326–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Liu, H.; Yuan, X.; Gao, K.; Duan, J. Comparative study of whole exome sequencing-based copy number variation detection tools. BMC Bioinform. 2020, 21, 97. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Qin, T.; Wang, X.; Li, J.; Lin, X.; Wang, D.; Lin, Z.; Zhang, X.; Li, X.; Lin, H.; et al. Whole-Exome Sequencing and Copy Number Analysis in a Patient with Warburg Micro Syndrome. Genes 2022, 13, 2364. https://doi.org/10.3390/genes13122364
Wang Q, Qin T, Wang X, Li J, Lin X, Wang D, Lin Z, Zhang X, Li X, Lin H, et al. Whole-Exome Sequencing and Copy Number Analysis in a Patient with Warburg Micro Syndrome. Genes. 2022; 13(12):2364. https://doi.org/10.3390/genes13122364
Chicago/Turabian StyleWang, Qiwei, Tingfeng Qin, Xun Wang, Jing Li, Xiaoshan Lin, Dongni Wang, Zhuoling Lin, Xulin Zhang, Xiaoyan Li, Haotian Lin, and et al. 2022. "Whole-Exome Sequencing and Copy Number Analysis in a Patient with Warburg Micro Syndrome" Genes 13, no. 12: 2364. https://doi.org/10.3390/genes13122364