
Two Novel Variants of WDR26 in Chinese Patients with Intellectual Disability


Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Genetic Studies and Variant Assessment
3. Results
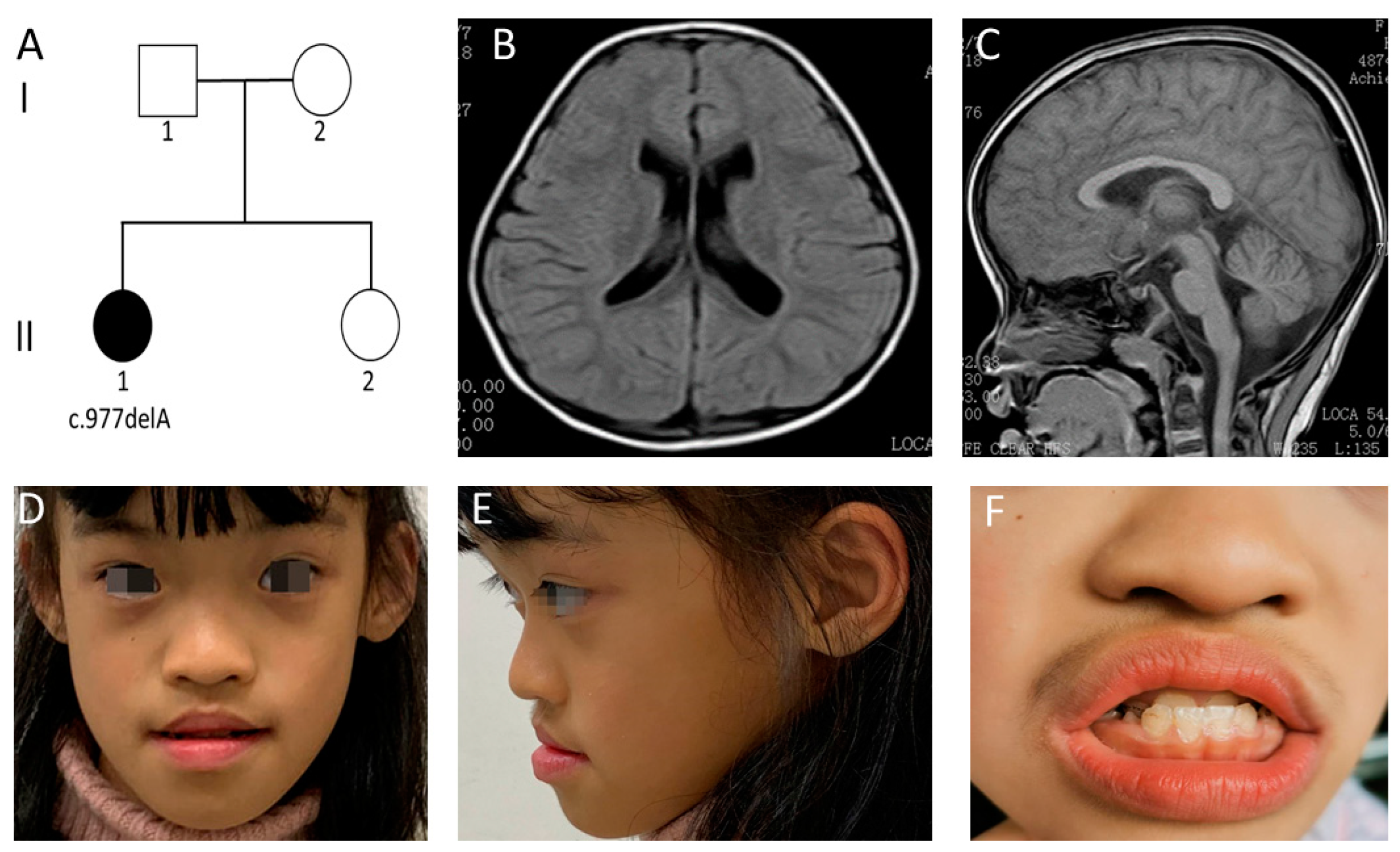
3.1. Clinical Features of Case 1
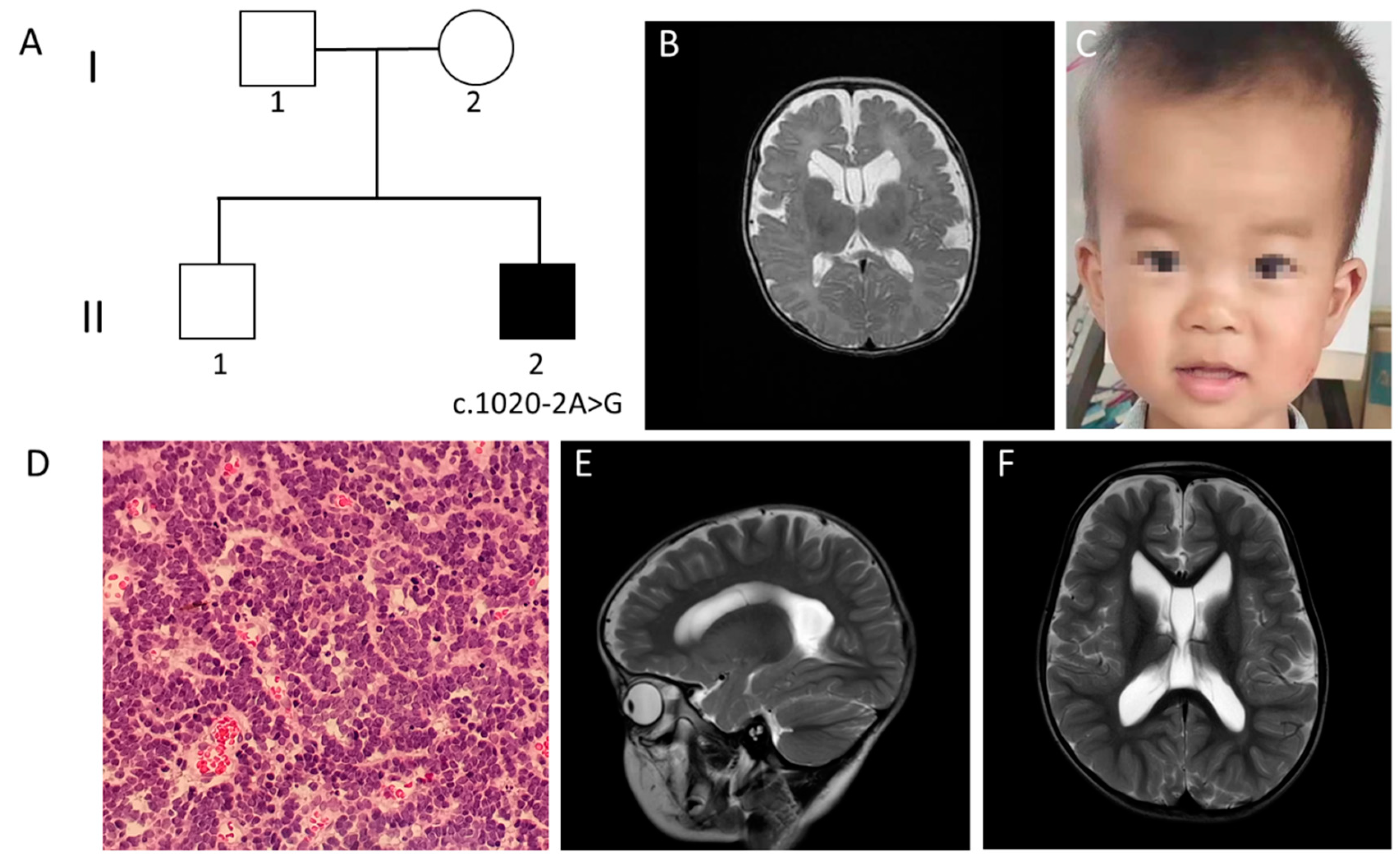
3.2. Clinical Features of Case 2
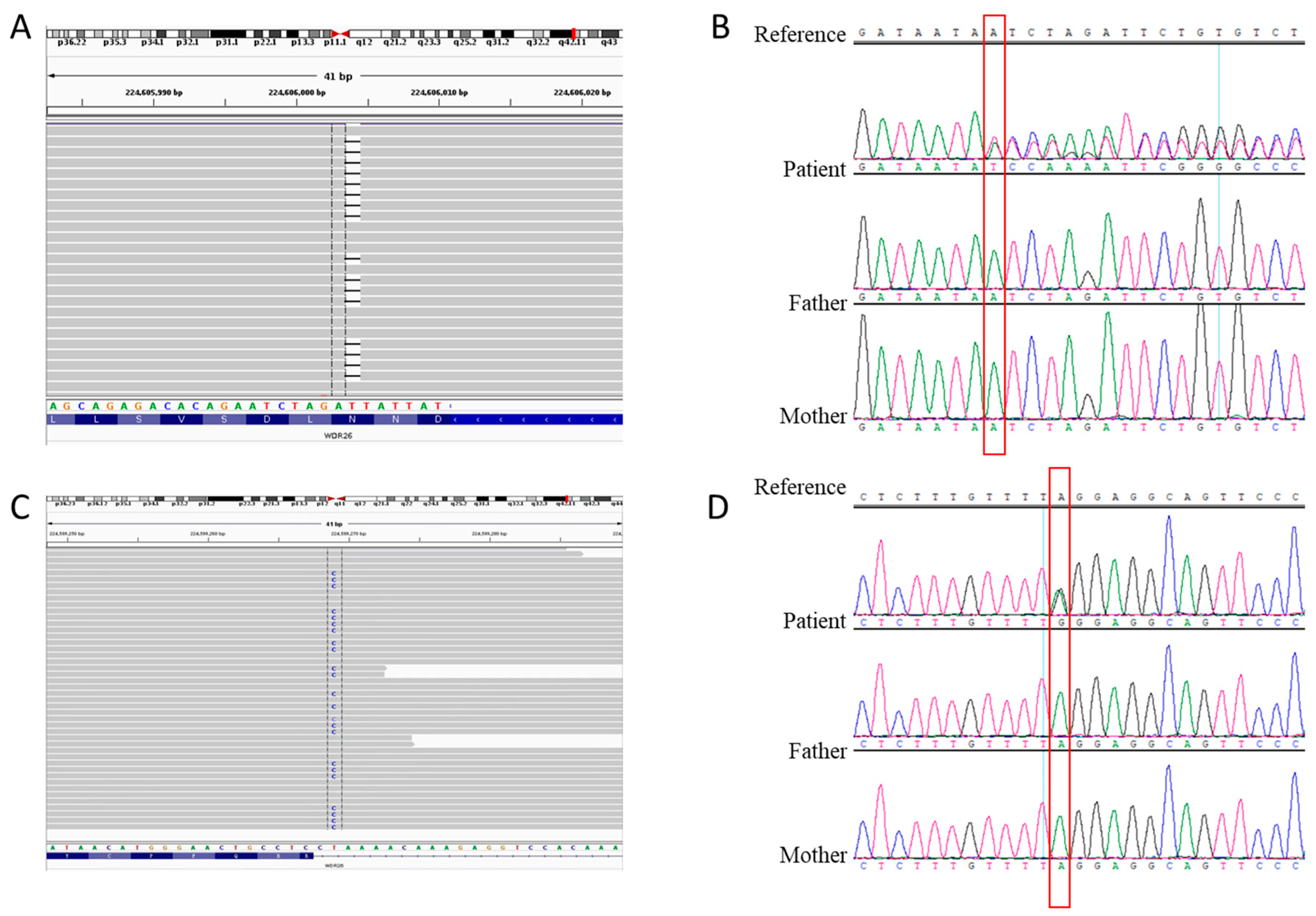
3.3. Genetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skraban, C.M.; Wells, C.F.; Markose, P.; Cho, M.T.; Nesbitt, A.I.; Au, P.Y.B.; Begtrup, A.; Bernat, J.A.; Bird, L.M.; Cao, K.; et al. WDR26 Haploinsufficiency Causes a Recognizable Syndrome of Intellectual Disability, Seizures, Abnormal Gait, and Distinctive Facial Features. Am. J. Hum. Genet. 2017, 101, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagishita, T.; Yamamoto-Shimojima, K.; Nakano, S.; Sasaki, T.; Shigematsu, H.; Imai, K.; Yamamoto, T. Phenotypic features of 1q41q42 microdeletion including WDR26 and FBXO28 are clinically recognizable: The first case from Japan. Brain Dev. 2019, 41, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, Y.; Wei, X.; Yuan, C.; Yuan, X.; Xiao, X. A novel WD-40 repeat protein WDR26 suppresses H2O2-induced cell death in neural cells. Neurosci. Lett. 2009, 460, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Zhen, R.; Moo, C.; Zhao, Z.; Chen, M.; Feng, H.; Zheng, X.; Zhang, L.; Shi, J.; Chen, C. Wdr26 regulates nuclear condensation in developing erythroblasts. Blood 2020, 135, 208–219. [Google Scholar] [CrossRef]
- Cospain, A.; Schaefer, E.; Faoucher, M.; Dubourg, C.; Carré, W.; Bizaoui, V.; Assoumani, J.; Van Maldergem, L.; Piton, A.; Gérard, B.; et al. Skraban-Deardorff syndrome: Six new cases of WDR26-related disease and expansion of the clinical phenotype. Clin. Genet. 2021, 99, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Pavinato, L.; Trajkova, S.; Grosso, E.; Giorgio, E.; Bruselles, A.; Radio, F.C.; Pippucci, T.; Dimartino, P.; Tartaglia, M.; Petlichkovski, A.; et al. Expanding the clinical phenotype of the ultra-rare Skraban-Deardorff syndrome: Two novel individuals with WDR26 loss-of-function variants and a literature review. Am. J. Med. Genet. A 2021, 185, 1712–1720. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, Q.; Sun, H.; Zhang, H.; Wang, R.; Li, S.; Sun, D.; Yang, X.A.; Jin, Y. The combination of whole-exome sequencing and copy number variation sequencing enables the diagnosis of rare neurological disorders. Clin. Genet. 2019, 96, 140–150. [Google Scholar] [CrossRef]
- Takata, A.; Miyake, N.; Tsurusaki, Y.; Fukai, R.; Miyatake, S.; Koshimizu, E.; Kushima, I.; Okada, T.; Morikawa, M.; Uno, Y.; et al. Integrative Analyses of De Novo Mutations Provide Deeper Biological Insights into Autism Spectrum Disorder. Cell Rep. 2018, 22, 734–747. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Hu, G.; Luo, J.; Fang, D.; Yu, Y.; Wang, X.; Chen, J.; Qiu, W. Mutations in methionyl-tRNA synthetase gene in a Chinese family with interstitial lung and liver disease, postnatal growth failure and anemia. J. Hum. Genet. 2017, 62, 647–651. [Google Scholar] [CrossRef]
- Sun, Y.; Ye, X.; Fan, Y.; Wang, L.; Luo, X.; Liu, H.; Gao, X.; Gong, Z.; Wang, Y.; Qiu, W.; et al. High Detection Rate of Copy Number Variations Using Capture Sequencing Data: A Retrospective Study. Clin. Chem. 2020, 66, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Shih, D.J.; Remke, M.; Cho, Y.J.; Kool, M.; Hawkins, C.; Eberhart, C.G.; Dubuc, A.; Guettouche, T.; Cardentey, Y.; et al. Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta Neuropathol. 2012, 123, 615–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Liu, Y.; Cai, W.; Ma, J.; Ni, K.; Chen, M.; Wang, C.; Liu, Y.; Zhu, Y.; Liu, Z.; et al. Detection of Disease-Causing SNVs/Indels and CNVs in Single Test Based on Whole Exome Sequencing: A Retrospective Case Study in Epileptic Encephalopathies. Front. Pediatr. 2021, 9, 635703. [Google Scholar] [CrossRef]
- Shaffer, L.G.; Theisen, A.; Bejjani, B.A.; Ballif, B.C.; Aylsworth, A.S.; Lim, C.; McDonald, M.; Ellison, J.W.; Kostiner, D.; Saitta, S.; et al. The discovery of microdeletion syndromes in the post-genomic era: Review of the methodology and characterization of a new 1q41q42 microdeletion syndrome. Genet. Med. 2007, 9, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Au, P.Y.; Argiropoulos, B.; Parboosingh, J.S.; Micheil Innes, A. Refinement of the critical region of 1q41q42 microdeletion syndrome identifies FBXO28 as a candidate causative gene for intellectual disability and seizures. Am. J. Med. Genet. A 2014, 164a, 441–448. [Google Scholar] [CrossRef]
- Cassina, M.; Rigon, C.; Casarin, A.; Vicenzi, V.; Salviati, L.; Clementi, M. FBXO28 is a critical gene of the 1q41q42 microdeletion syndrome. Am. J. Med. Genet. A 2015, 167, 1418–1420. [Google Scholar] [CrossRef]
- Chen, C.P.; Chern, S.R.; Wu, P.S.; Chen, S.W.; Wu, F.T.; Wang, W. Molecular cytogenetic characterization of a de novo chromosome 1q41-q42.11 microdeletion of paternal origin in a 15-year-old boy with mental retardation, developmental delay, autism and congenital heart defects. Taiwan J. Obstet. Gynecol. 2021, 60, 341–344. [Google Scholar] [CrossRef]
- Christensen, R.D.; Yaish, H.M. A neonate with the Pelger-Huët anomaly, cleft lip and palate, and agenesis of the corpus callosum, with a chromosomal microdeletion involving 1q41 to 1q42.12. J. Perinatol. 2012, 32, 238–240. [Google Scholar] [CrossRef] [Green Version]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Xie, Y.; Kong, S.; Qiu, W.; Wang, X.; Wang, D.; Sun, X.; Sun, D. Psychomotor retardation with a 1q42.11-q42.12 deletion. Hereditas 2017, 154, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarci, S.; Ackerman, K.G.; Russell, M.K.; Longoni, M.; Sougnez, C.; Noonan, K.M.; Hatchwell, E.; Zhang, X.; Pieretti Vanmarcke, R.; Anyane-Yeboa, K.; et al. Characterization of the chromosome 1q41q42.12 region, and the candidate gene DISP1, in patients with CDH. Am. J. Med. Genet. A 2010, 152a, 2493–2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzeu, J.F.; Vianna-Morgante, A.M.; Krepischi, A.C.; Oudakker, A.; Rosenberg, C.; Szuhai, K.; McGill, J.; Maccraughan, J.; van Bokhoven, H.; Brunner, H.G. Deletions encompassing 1q41q42.1 and clinical features of autosomal dominant Robinow syndrome. Clin. Genet. 2010, 77, 404–407. [Google Scholar] [CrossRef]
- Rice, G.M.; Qi, Z.; Selzer, R.; Richmond, T.; Thompson, K.; Pauli, R.M.; Yu, J. Microdissection-based high-resolution genomic array analysis of two patients with cytogenetically identical interstitial deletions of chromosome 1q but distinct clinical phenotypes. Am. J. Med. Genet. A 2006, 140, 1637–1643. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, J.A.; Lacassie, Y.; El-Khechen, D.; Escobar, L.F.; Reggin, J.; Heuer, C.; Chen, E.; Jenkins, L.S.; Collins, A.T.; Zinner, S.; et al. New cases and refinement of the critical region in the 1q41q42 microdeletion syndrome. Eur. J. Med. Genet. 2011, 54, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Slavotinek, A.M.; Moshrefi, A.; Lopez Jiminez, N.; Chao, R.; Mendell, A.; Shaw, G.M.; Pennacchio, L.A.; Bates, M.D. Sequence variants in the HLX gene at chromosome 1q41-1q42 in patients with diaphragmatic hernia. Clin. Genet. 2009, 75, 429–439. [Google Scholar] [CrossRef] [Green Version]
- Spreiz, A.; Haberlandt, E.; Baumann, M.; Baumgartner Sigl, S.; Fauth, C.; Gautsch, K.; Karall, D.; Janetschek, C.; Rostasy, K.; Scholl-Bürgi, S.; et al. Chromosomal microaberrations in patients with epilepsy, intellectual disability, and congenital anomalies. Clin. Genet. 2014, 86, 361–366. [Google Scholar] [CrossRef]
- Wat, M.J.; Veenma, D.; Hogue, J.; Holder, A.M.; Yu, Z.; Wat, J.J.; Hanchard, N.; Shchelochkov, O.A.; Fernandes, C.J.; Johnson, A.; et al. Genomic alterations that contribute to the development of isolated and non-isolated congenital diaphragmatic hernia. J. Med. Genet. 2011, 48, 299–307. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Features | Patient 1 | Patient 2 | WDR26 Variants [1,5,6] (n = 23) | Chromosome 1q41–q42 Deletion Syndrome [1,2,11,12,13,14,15,16,17,18,19,20,21,22,23,24] (n = 29) |
|---|---|---|---|---|
| Developmental delay or intellectual disability (ID) | + | + | 23/23 | 27/27 |
| Limited speech | + | + | 23/23 | 8/8 |
| Seizures | – | – | 22/23 | 17/27 |
| CNS structural anomalies | + | + | 15/22 | 18/24 |
| Hypotonia | + | + | 14/20 | 9/21 |
| Abnormal gait | + | + | 14/17 | 3/16 |
| Happy and/or friendly personality | + | + | 18/19 | 4/4 |
| Autistic and/or repetitive behaviors or posturing | + | – | 11/17 | 3/3 |
| Facial features | ||||
| Coarse facial features | + | + | 15/23 | 18/22 |
| Full cheeks as a child Large irises or rounded/short/slanting palpebral fissures | – + (rounded) | + + (rounded) | 18/21 15/23 (rounded) | 9/10 7/12 (slanting) |
| Abnormal eyebrows | – | – | 11/23 | 6/13 |
| Depressed nasal root | + | + | 12/23 | 18/22 |
| Anteverted nares | + | – | 15/23 | 12/16 |
| Full nasal tip | + | + | 19/23 | 14/20 |
| Prominent maxilla | – | – | 16/23 | 10/17 |
| Protruding or full, tented upper lip | – | – | 16/23 | 13/18 |
| Wide mouth | + | – | 12/17 | 8/17 |
| Decreased cupid’s bow | + | – | 11/23 | 12/15 |
| Widely spaced teeth | + | – | 18/21 | 9/10 |
| Abnormal gums | + | + | 13/19 | 8/9 |
| Hypertelorism | – | + | 5/15 | 13/21 |
| Ophthalmologic abnormalities | – | – | 10/17 | 3/8 |
| Nail hypoplasia | – | – | 3/13 | 6/6 |
| Short stature | – | – | 3/21 | 5/19 |
| Digit abnormalities | – | – | 5/14 | 8/10 |
| GI difficulties | + | – | 11/16 | 7/8 |
| Orthopaedic disorders | – | – | 13/17 | 15/15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, J.; Xu, M.; Zhu, X.; Zhang, Y. Two Novel Variants of WDR26 in Chinese Patients with Intellectual Disability. Genes 2022, 13, 813. https://doi.org/10.3390/genes13050813
Hu J, Xu M, Zhu X, Zhang Y. Two Novel Variants of WDR26 in Chinese Patients with Intellectual Disability. Genes. 2022; 13(5):813. https://doi.org/10.3390/genes13050813
Chicago/Turabian StyleHu, Jiacheng, Mingming Xu, Xiaobo Zhu, and Yu Zhang. 2022. "Two Novel Variants of WDR26 in Chinese Patients with Intellectual Disability" Genes 13, no. 5: 813. https://doi.org/10.3390/genes13050813