
New Insights into the Neuromyogenic Spectrum of a Gain of Function Mutation in SPTLC1

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Immortalized Lymphoblastoid Cells
2.2. Proteomic Profiling of Immortalized Lymphoblastoid Cells
2.2.1. Sample Preparation for Mass Spectrometry
2.2.2. Proteomic Analysis
2.2.3. Data Analysis
2.2.4. In Silico Studies of Proteomic Findings
2.3. Morphological Analysis of Quadriceps Muscle
2.4. Transcript Studies on Quadriceps Muscle
2.5. Coherent Anti-Stokes Raman Scattering (CARS) and Statistical Evaluation of Muscle Fiber Calibres
2.6. SPTLC1 mRNA Expression and Transcript Studies
3. Results
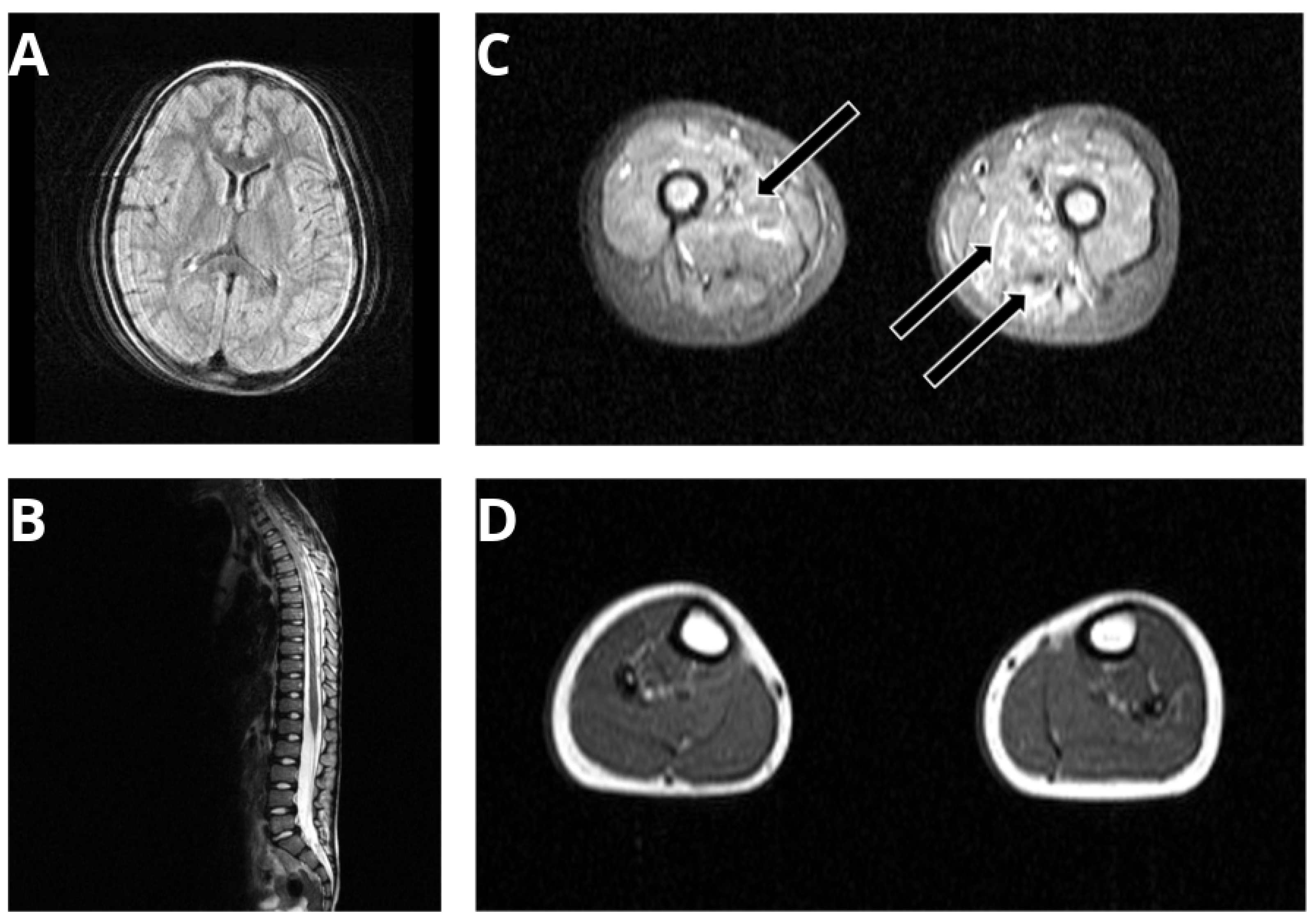
3.1. Phenotyping of a SPTLC1-Related ALS Case with Juvenile Onset
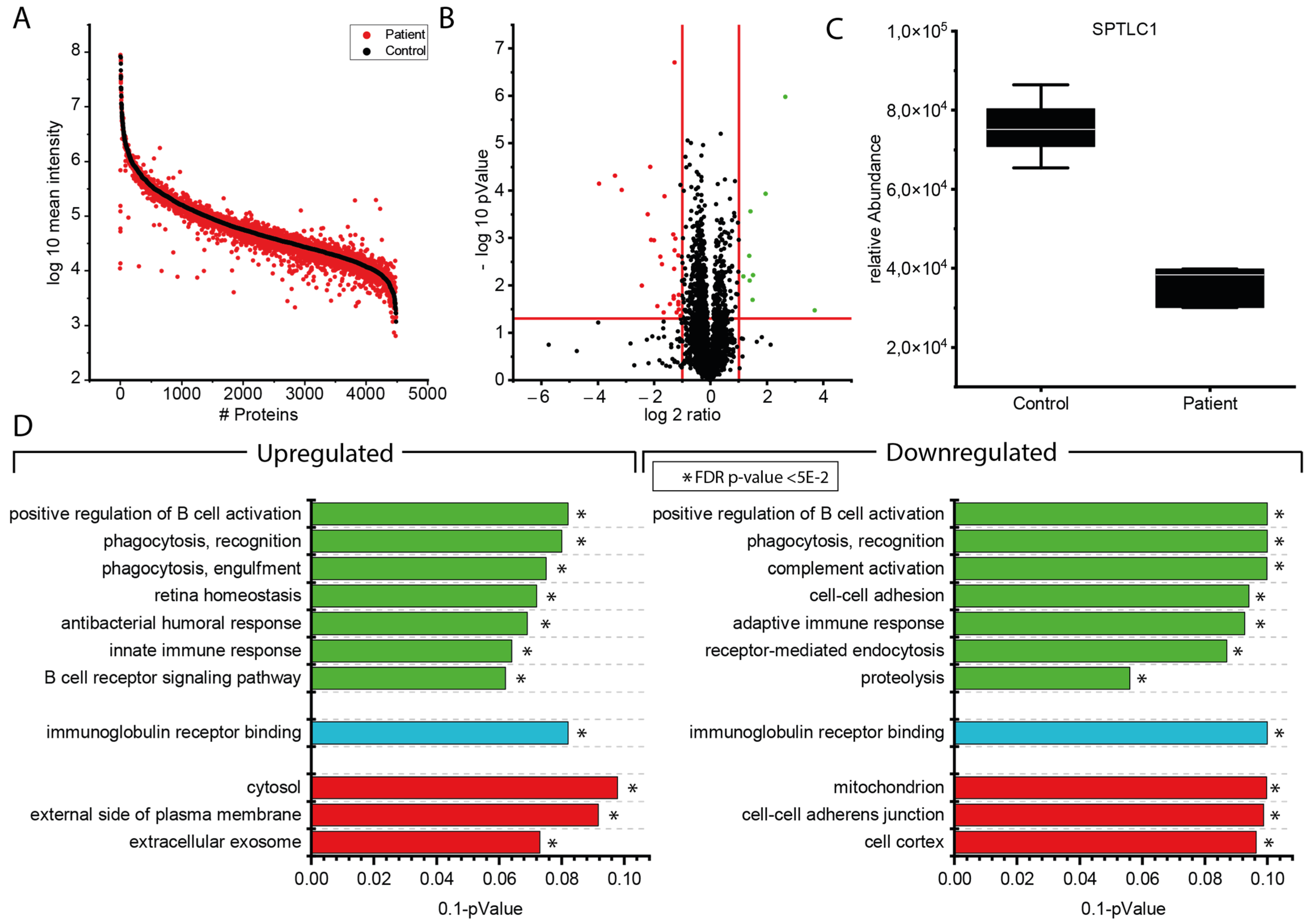
3.2. Proteomic Findings in Lymphoblastoid Cells Derived from the p.(A20S)-SPTLC1-Patient
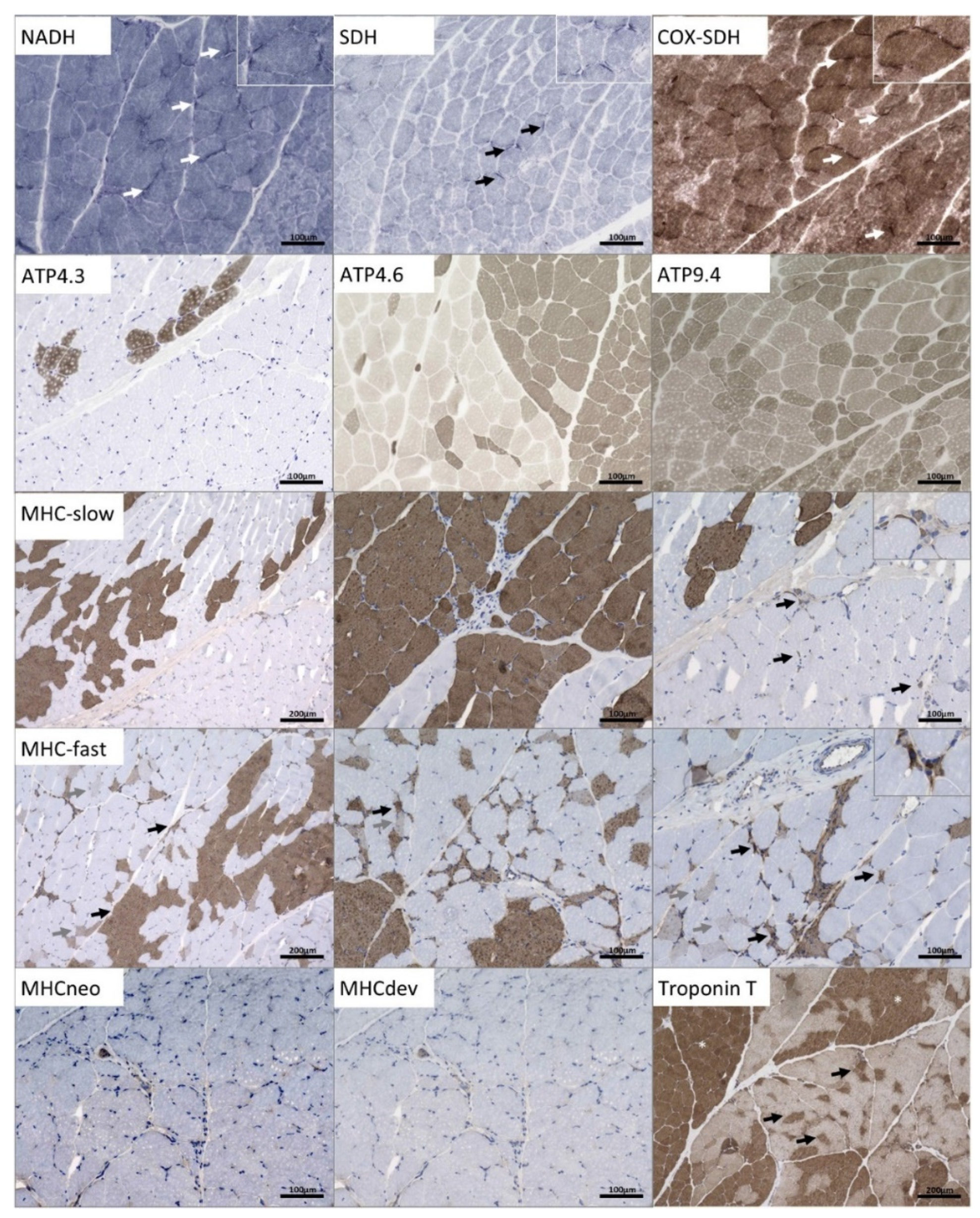
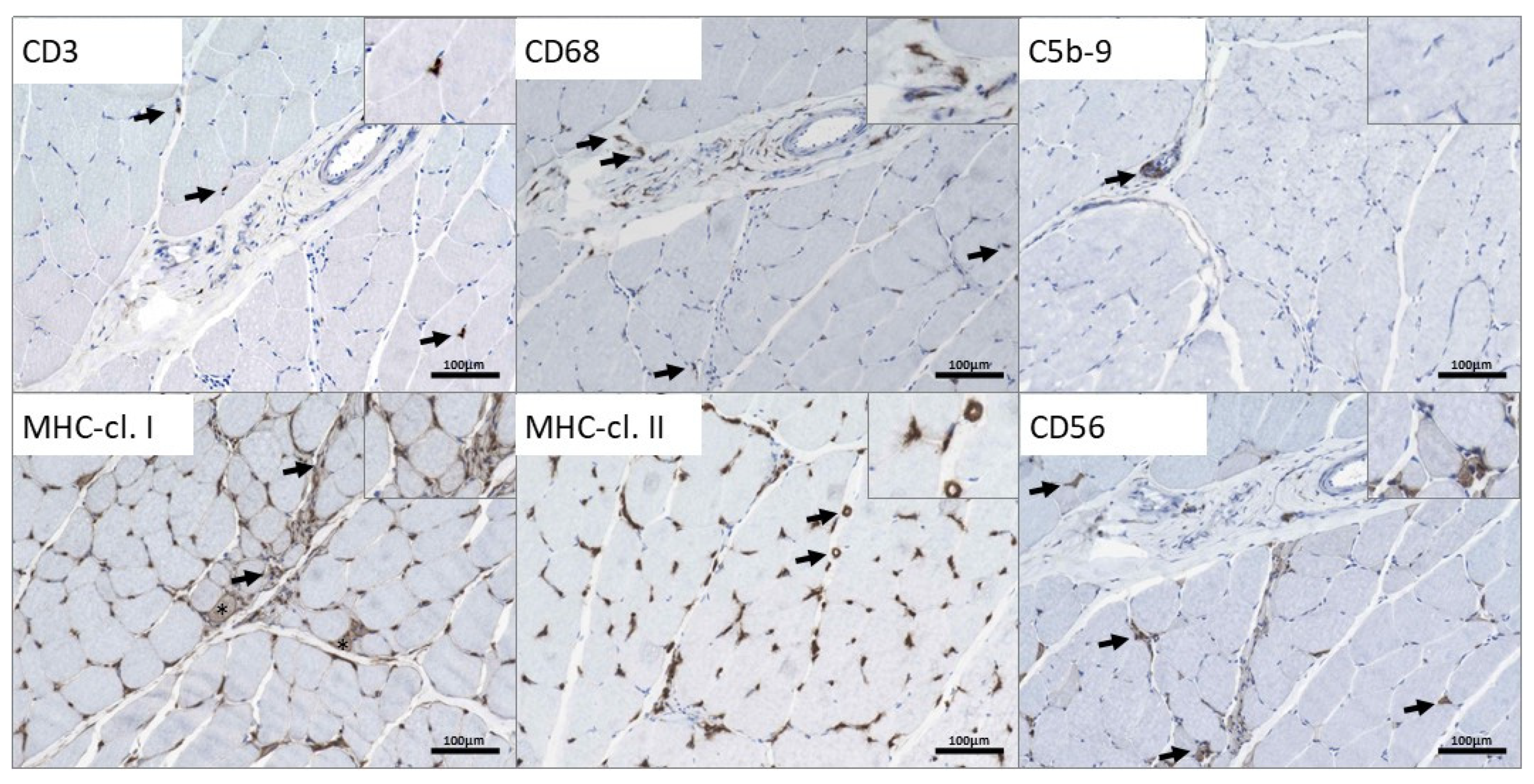
3.3. Microscopic Findings on a Quadriceps Biopsy Derived from the p.(A20S)-SPTLC1-Patient
Coherent Anti-Stokes Raman Scattering (CARS) Is a Nonlinear Variant of the Raman Effect
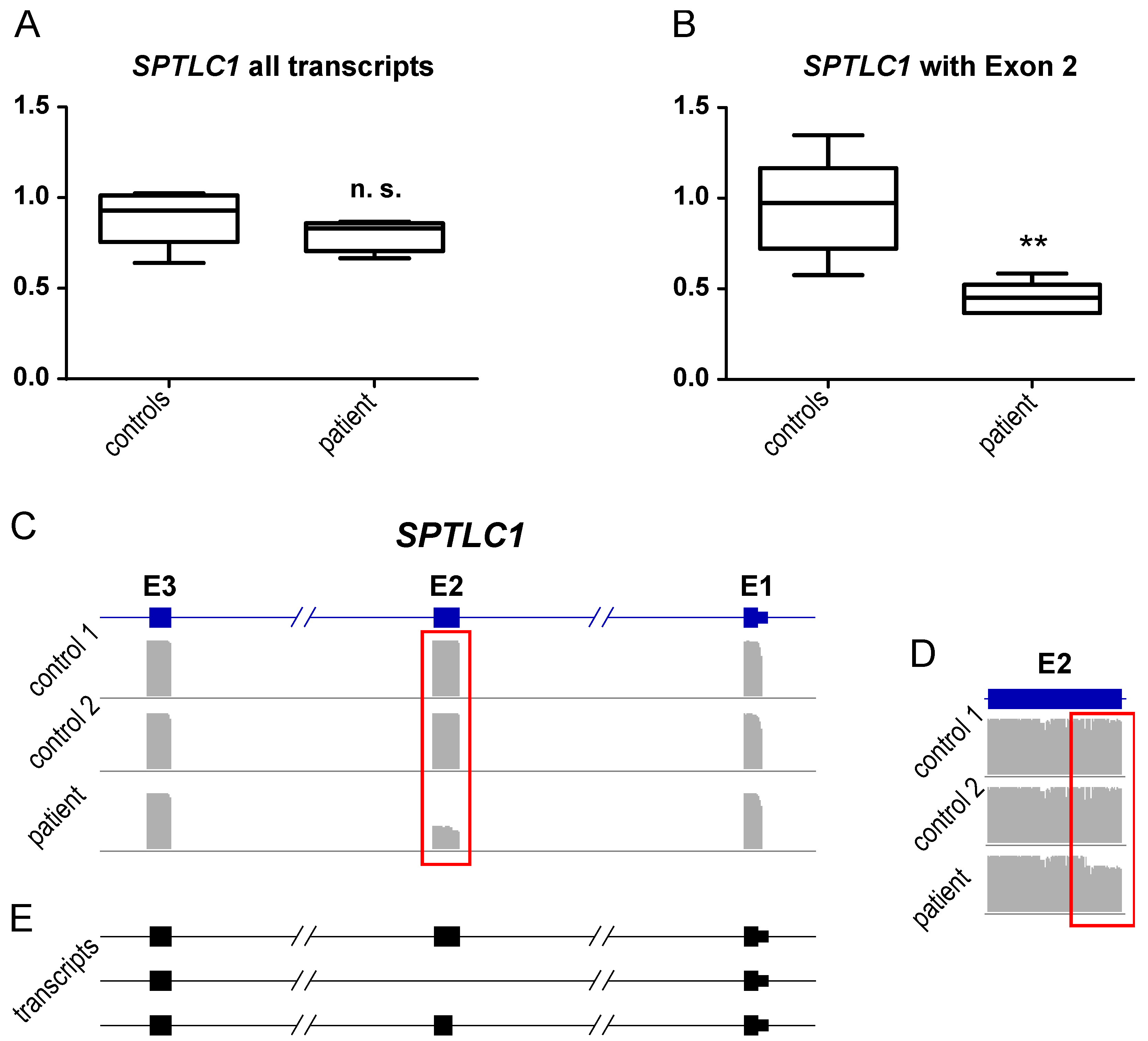
3.4. Transcript Findings in Quadriceps Muscle Derived from the p.(A20S)-SPTLC1-Patient
4. Discussion
4.1. Clinical and Microscopic Findings of the p.(A20S)-SPTLC1-Patient
4.2. Molecular Signature of p.(A20S)-SPTLC1-Mutant Lymphoblastoid Cells
4.3. Findings in the Quadriceps Biopsy of the p.(A20S)-SPTLC1-Patient
5. Conclusions
- First manifestation of proximal muscle weakness should be considered in the clinical diagnosis of SPTLC1-related juvenile ALS
- P.(A20S)-SPTLC1-related ALS leads to muscle denervation accompanied by fiber-type grouping, subsarcolemmal accumulation of mitochondria in type I fibers, and mild inflammation
- Aggregates of proteins or lipids were not identified in patient-derived quadriceps muscle by CARS microscopy
- Proteomic profiling on p.(A20S)-SPTLC1-mutant lymphoblastoid cells unraveled a variety of proteins with potential impact on clinical manifestation of symptoms of SPTLC1-related phenotypical spectrum
- Further functional in vitro studies are needed to systematically address the impact of these proteins in the pathogenesis of increased SPTLC1 function
- The molecular genetic SPTLC1 variant identified in our patient is associated with altered splicing and decrease of protein level
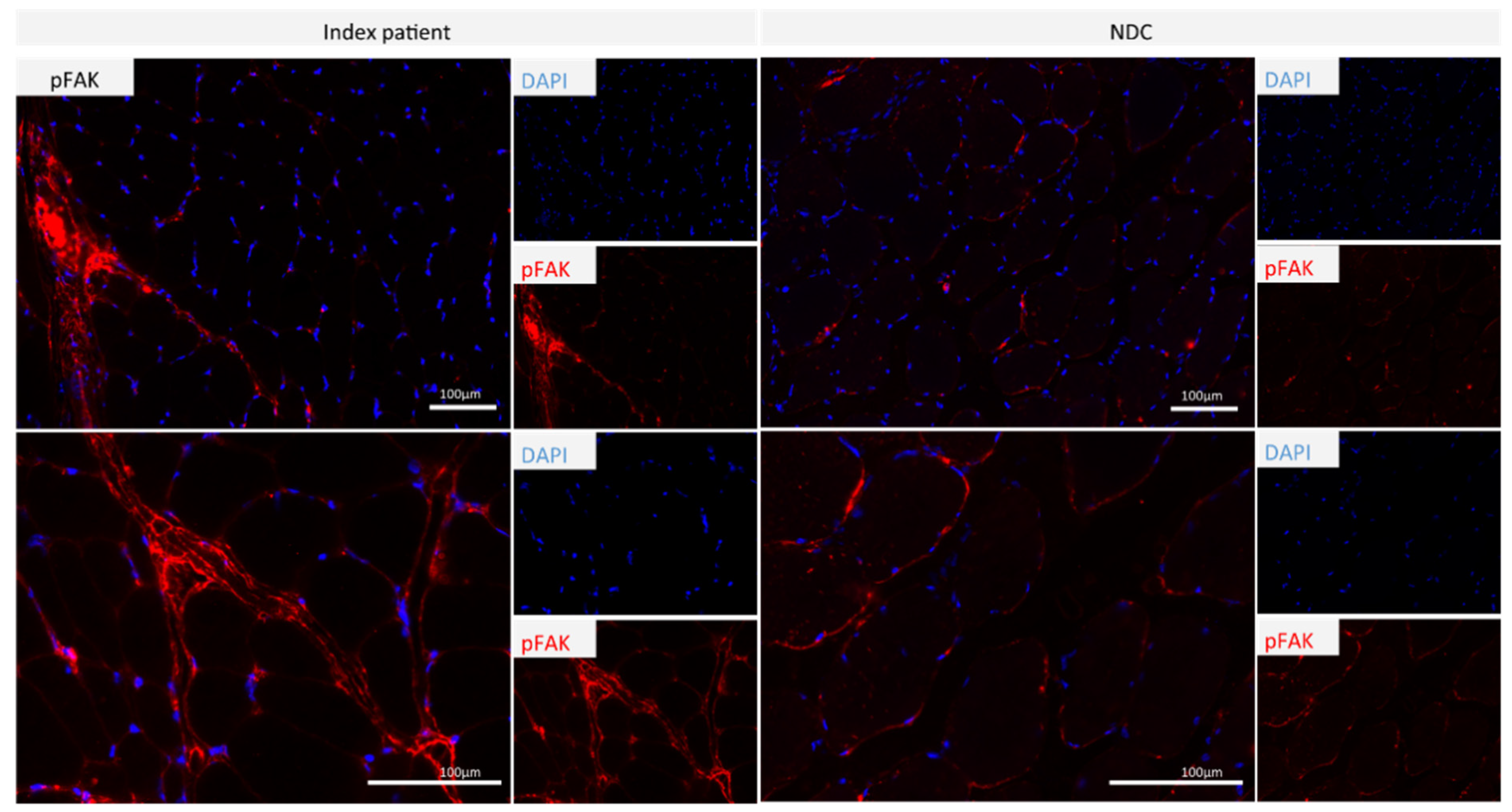
- Increased expression of phosphor-FAK1 in patient-derived quadriceps muscle might contribute to fibrotic degeneration
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Han, G.; Gupta, S.D.; Gable, K.; Niranjanakumari, S.; Moitra, P.; Eichler, F.; Brown, R.H., Jr.; Harmon, J.M.; Dunn, T.M. Identification of small subunits of mammalian serine palmitoyltransferase that confer distinct acyl-CoA substrate specificities. Proc. Natl. Acad. Sci. USA 2009, 106, 8186–8191. [Google Scholar] [CrossRef] [Green Version]
- Myers, S.J.; Malladi, C.S.; Hyland, R.A.; Bautista, T.; Boadle, R.; Robinson, P.J.; Nicholson, G.A. Mutations in the SPTLC1 protein cause mitochondrial structural abnormalities and endoplasmic reticulum stress in lymphoblasts. DNA Cell Biol. 2014, 33, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Auer-Grumbach, M.; De Jonghe, P.; Verhoeven, K.; Timmerman, V.; Wagner, K.; Hartung, H.P.; Nicholson, G.A. Autosomal dominant inherited neuropathies with prominent sensory loss and mutilations: A review. Arch. Neurol. 2003, 60, 329–334. [Google Scholar] [CrossRef]
- Nicholson, G.A. SPTLC1-Related Hereditary Sensory Neuropathy. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Jennings, M.J.; Lochmüller, A.; Atalaia, A.; Horvath, R. Targeted Therapies for Hereditary Peripheral Neuropathies: Systematic Review and Steps Towards a ‘treatabolome’. J. Neuromuscul. Dis. 2021, 8, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Mohassel, P.; Donkervoort, S.; Lone, M.A.; Nalls, M.; Gable, K.; Gupta, S.D.; Foley, A.R.; Hu, Y.; Saute, J.A.M.; Moreira, A.L.; et al. Childhood amyotrophic lateral sclerosis caused by excess sphingolipid synthesis. Nat. Med. 2021, 27, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Chia, R.; Miller, D.E.; Li, R.; Kumaran, R.; Abramzon, Y.; Alahmady, N.; Renton, A.E.; Topp, S.D.; Gibbs, J.R.; et al. Association of Variants in the SPTLC1 Gene With Juvenile Amyotrophic Lateral Sclerosis. JAMA Neurol. 2021, 78, 1236–1248. [Google Scholar] [CrossRef]
- Burkhart, J.M.; Schumbrutzki, C.; Wortelkamp, S.; Sickmann, A.; Zahedi, R.P. Systematic and quantitative comparison of digest efficiency and specificity reveals the impact of trypsin quality on MS-based proteomics. J. Proteom. 2012, 75, 1454–1462. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Kölbel, H.; Preuße, C.; Brand, L.; von Moers, A.; Della Marina, A.; Schuelke, M.; Roos, A.; Goebel, H.H.; Schara-Schmidt, U.; Stenzel, W. Inflammation, fibrosis and skeletal muscle regeneration in LGMDR9 are orchestrated by macrophages. Neuropathol. Appl. Neurobiol. 2021, 47, 856–866. [Google Scholar] [CrossRef]
- Roos, A.; Preusse, C.; Hathazi, D.; Goebel, H.H.; Stenzel, W. Proteomic Profiling Unravels a Key Role of Specific Macrophage Subtypes in Sporadic Inclusion Body Myositis. Front. Immunol. 2019, 10, 1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentschel, A.; Czech, A.; Münchberg, U.; Freier, E.; Schara-Schmidt, U.; Sickmann, A.; Reimann, J.; Roos, A. Protein signature of human skin fibroblasts allows the study of the molecular etiology of rare neurological diseases. Orphanet J. Rare Dis. 2021, 16, 73. [Google Scholar] [CrossRef] [PubMed]
- Töpf, A.; Johnson, K.; Bates, A.; Phillips, L.; Chao, K.R.; England, E.M.; Laricchia, K.M.; Mullen, T.; Valkanas, E.; Xu, L.; et al. Sequential targeted exome sequencing of 1001 patients affected by unexplained limb-girdle weakness. Genet. Med. Off. J. Am. Coll. Med. Genet. 2020, 22, 1478–1488. [Google Scholar] [CrossRef]
- Kollipara, L.; Buchkremer, S.; Coraspe, J.A.G.; Hathazi, D.; Senderek, J.; Weis, J.; Zahedi, R.P.; Roos, A. In-depth phenotyping of lymphoblastoid cells suggests selective cellular vulnerability in Marinesco-Sjögren syndrome. Oncotarget 2017, 8, 68493–68516. [Google Scholar] [CrossRef] [Green Version]
- Marshall, L.L.; Stimpson, S.E.; Hyland, R.; Coorssen, J.R.; Myers, S.J. Increased lipid droplet accumulation associated with a peripheral sensory neuropathy. J. Chem. Biol. 2014, 7, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Rinia, H.A.; Burger, K.N.; Bonn, M.; Müller, M. Quantitative label-free imaging of lipid composition and packing of individual cellular lipid droplets using multiplex CARS microscopy. Biophys. J. 2008, 95, 4908–4914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.X.; Xie, X.S. Vibrational spectroscopic imaging of living systems: An emerging platform for biology and medicine. Science 2015, 350, aaa8870. [Google Scholar] [CrossRef]
- van der Kooi, A.J.; Ginjaar, H.B.; Busch, H.F.; Wokke, J.H.; Barth, P.G.; de Visser, M. Limb girdle muscular dystrophy: A pathological and immunohistochemical reevaluation. Muscle Nerve 1998, 21, 584–590. [Google Scholar] [CrossRef]
- Nadeau, A.; Kinali, M.; Main, M.; Jimenez-Mallebrera, C.; Aloysius, A.; Clement, E.; North, B.; Manzur, A.Y.; Robb, S.A.; Mercuri, E.; et al. Natural history of Ullrich congenital muscular dystrophy. Neurology 2009, 73, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Roos, A.; Thompson, R.; Horvath, R.; Lochmüller, H.; Sickmann, A. Intersection of Proteomics and Genomics to “Solve the Unsolved” in Rare Disorders such as Neurodegenerative and Neuromuscular Diseases. Proteom. Clin. Appl. 2018, 12, 1700073. [Google Scholar] [CrossRef]
- Thompson, R.; Spendiff, S.; Roos, A.; Bourque, P.R.; Warman Chardon, J.; Kirschner, J.; Horvath, R.; Lochmüller, H. Advances in the diagnosis of inherited neuromuscular diseases and implications for therapy development. Lancet Neurol. 2020, 19, 522–532. [Google Scholar] [CrossRef]
- Juneja, M.; Azmi, A.; Baets, J.; Roos, A.; Jennings, M.J.; Saveri, P.; Pisciotta, C.; Bernard-Marissal, N.; Schneider, B.L.; Verfaillie, C.; et al. PFN2 and GAMT as common molecular determinants of axonal Charcot-Marie-Tooth disease. J. Neurol. Neurosurg. Psychiatry 2018, 89, 870–878. [Google Scholar] [CrossRef]
- Auer-Grumbach, M.; Bode, H.; Pieber, T.R.; Schabhüttl, M.; Fischer, D.; Seidl, R.; Graf, E.; Wieland, T.; Schuh, R.; Vacariu, G.; et al. Mutations at Ser331 in the HSN type I gene SPTLC1 are associated with a distinct syndromic phenotype. Eur. J. Med. Genet. 2013, 56, 266–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurklinsky, S.; Chen, J.; McNiven, M.A. Growth cone morphology and spreading are regulated by a dynamin-cortactin complex at point contacts in hippocampal neurons. J. Neurochem. 2011, 117, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, G.; Kim, E.J.; Cicvaric, A.; Sase, S.; Gröger, M.; Höger, H.; Sialana, F.J.; Berger, J.; Monje, F.J.; Lubec, G. Drebrin depletion alters neurotransmitter receptor levels in protein complexes, dendritic spine morphogenesis and memory-related synaptic plasticity in the mouse hippocampus. J. Neurochem. 2015, 134, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz, R.; Berberich, S.; Rathgeber, L.; Kolleker, A.; Köhr, G.; Kornau, H.C. AMPA receptor signaling through BRAG2 and Arf6 critical for long-term synaptic depression. Neuron 2010, 66, 768–780. [Google Scholar] [CrossRef] [Green Version]
- Tedeschi, V.; Petrozziello, T.; Secondo, A. Ca2+ dysregulation in the pathogenesis of amyotrophic lateral sclerosis. Int. Rev. Cell Mol. Biol. 2021, 363, 21–47. [Google Scholar] [CrossRef]
- Gregory, J.M.; Livesey, M.R.; McDade, K.; Selvaraj, B.T.; Barton, S.K.; Chandran, S.; Smith, C. Dysregulation of AMPA receptor subunit expression in sporadic ALS post-mortem brain. J. Pathol. 2020, 250, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Rico, B.; Beggs, H.E.; Schahin-Reed, D.; Kimes, N.; Schmidt, A.; Reichardt, L.F. Control of axonal branching and synapse formation by focal adhesion kinase. Nat. Neurosci. 2004, 7, 1059–1069. [Google Scholar] [CrossRef]
- Tu, H.; Xu, C.; Zhang, W.; Liu, Q.; Rondard, P.; Pin, J.P.; Liu, J. GABAB receptor activation protects neurons from apoptosis via IGF-1 receptor transactivation. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 749–759. [Google Scholar] [CrossRef]
- de Oliveira, A.S.; Santiago, F.E.; Balioni, L.F.; Ferrari Mde, F.; Almeida, M.C.; Carrettiero, D.C. BAG2 expression dictates a functional intracellular switch between the p38-dependent effects of nicotine on tau phosphorylation levels via the α7 nicotinic receptor. Exp. Neurol. 2016, 275 Pt 1, 69–77. [Google Scholar] [CrossRef]
- Qu, D.; Hage, A.; Don-Carolis, K.; Huang, E.; Joselin, A.; Safarpour, F.; Marcogliese, P.C.; Rousseaux, M.W.; Hewitt, S.J.; Huang, T.; et al. BAG2 Gene-mediated Regulation of PINK1 Protein Is Critical for Mitochondrial Translocation of PARKIN and Neuronal Survival. J. Biol. Chem. 2015, 290, 30441–30452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stimpson, S.E.; Coorssen, J.R.; Myers, S.J. Mitochondrial protein alterations in a familial peripheral neuropathy caused by the V144D amino acid mutation in the sphingolipid protein, SPTLC1. J. Chem. Biol. 2015, 8, 25–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, H.; Schneider, M.; Williamson, D.B.; Ito, A.; Takeuchi, M.; Handford, P.A.; Haltiwanger, R.S. Two novel protein O-glucosyltransferases that modify sites distinct from POGLUT1 and affect Notch trafficking and signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E8395–E8402. [Google Scholar] [CrossRef] [Green Version]
- Cui, M.; Ying, R.; Jiang, X.; Li, G.; Zhang, X.; Zheng, J.; Tam, K.Y.; Liang, B.; Shi, A.; Göbel, V.; et al. A Model of Hereditary Sensory and Autonomic Neuropathy Type 1 Reveals a Role of Glycosphingolipids in Neuronal Polarity. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 5816–5834. [Google Scholar] [CrossRef] [Green Version]
- Epand, R.M.; Kam, A.; Bridgelal, N.; Saiga, A.; Topham, M.K. The alpha isoform of diacylglycerol kinase exhibits arachidonoyl specificity with alkylacylglycerol. Biochemistry 2004, 43, 14778–14783. [Google Scholar] [CrossRef]
- Lung, M.; Shulga, Y.V.; Ivanova, P.T.; Myers, D.S.; Milne, S.B.; Brown, H.A.; Topham, M.K.; Epand, R.M. Diacylglycerol kinase epsilon is selective for both acyl chains of phosphatidic acid or diacylglycerol. J. Biol. Chem. 2009, 284, 31062–31073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulga, Y.V.; Topham, M.K.; Epand, R.M. Substrate specificity of diacylglycerol kinase-epsilon and the phosphatidylinositol cycle. FEBS Lett. 2011, 585, 4025–4028. [Google Scholar] [CrossRef] [Green Version]
- Shulga, Y.V.; Topham, M.K.; Epand, R.M. Study of arachidonoyl specificity in two enzymes of the PI cycle. J. Mol. Biol. 2011, 409, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Liu, K.; Sasaki, S.; Kunii, N.; Sakai, H.; Mizuno, H.; Saga, H.; Sakane, F. Evaluations of the selectivities of the diacylglycerol kinase inhibitors R59022 and R59949 among diacylglycerol kinase isozymes using a new non-radioactive assay method. Pharmacology 2013, 92, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Sevinsky, C.J.; Khan, F.; Kokabee, L.; Darehshouri, A.; Maddipati, K.R.; Conklin, D.S. NDRG1 regulates neutral lipid metabolism in breast cancer cells. Breast Cancer Res. 2018, 20, 55. [Google Scholar] [CrossRef] [PubMed]
- Li, L.X.; Liu, G.L.; Liu, Z.J.; Lu, C.; Wu, Z.Y. Identification and functional characterization of two missense mutations in NDRG1 associated with Charcot-Marie-Tooth disease type 4D. Hum. Mutat. 2017, 38, 1569–1578. [Google Scholar] [CrossRef]
- Gantner, M.L.; Eade, K.; Wallace, M.; Handzlik, M.K.; Fallon, R.; Trombley, J.; Bonelli, R.; Giles, S.; Harkins-Perry, S.; Heeren, T.F.C.; et al. Serine and Lipid Metabolism in Macular Disease and Peripheral Neuropathy. N. Engl. J. Med. 2019, 381, 1422–1433. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xue, P.; Chen, H.; Yuan, M.; Kang, Y.; Duscher, D.; Machens, H.G.; Chen, Z. Denervation drives skeletal muscle atrophy and induces mitochondrial dysfunction, mitophagy and apoptosis via miR-142a-5p/MFN1 axis. Theranostics 2020, 10, 1415–1432. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S. Muscle fiber type diversity revealed by anti-myosin heavy chain antibodies. FEBS J. 2018, 285, 3688–3694. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.; Bastide, B.; Maurage, C.A.; Dupont, E.; Montel, V.; Cieniewski-Bernard, C.; Cuisset, J.M.; Vallée, L.; Mounier, Y. Childhood spinal muscular atrophy induces alterations in contractile and regulatory protein isoform expressions. Neuropathol. Appl. Neurobiol. 2008, 34, 659–670. [Google Scholar] [CrossRef]
- Lyon, M.S.; Wosiski-Kuhn, M.; Gillespie, R.; Caress, J.; Milligan, C. Inflammation, Immunity, and amyotrophic lateral sclerosis: I. Etiology and pathology. Muscle Nerve 2019, 59, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Graham, Z.A.; Gallagher, P.M.; Cardozo, C.P. Focal adhesion kinase and its role in skeletal muscle. J. Muscle Res. Cell Motil. 2015, 36, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Kathiriya, J.J.; Nakra, N.; Nixon, J.; Patel, P.S.; Vaghasiya, V.; Alhassani, A.; Tian, Z.; Allen-Gipson, D.; Davé, V. Galectin-1 inhibition attenuates profibrotic signaling in hypoxia-induced pulmonary fibrosis. Cell Death Discov. 2017, 3, 17010. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Accession | Gene Name | Protein Name | Unique Peptides | Fold of Decrease | p-Value | Neurological Disease | OMIM |
|---|---|---|---|---|---|---|---|
| Q9HA77 | CARS2 | Probable cysteine-tRNA ligase, mitochondrial | 9 | 0.54 | 0.000 | Combined oxidative phosphorylation deficiency 27 | COXPD27; MIM:616672 |
| Q9NVH6 | TMLHE | Trimethyllysine dioxygenase, mitochondrial | 2 | 0.54 | 0.004 | Autism, X-linked 6 | AUTSX6; MIM:300872 |
| Q9ULJ6 | ZMIZ1 | Zinc finger MIZ domain-containing protein 1 | 2 | 0.51 | 0.019 | Neurodevelopmental disorder with dysmorphic facies and distal skeletal anomalies (NEDDFSA; MIM:618659) | NEDDFSA; MIM:618659 |
| Q92643 | PIGK | GPI-anchor transamidase | 2 | 0.50 | 0.016 | Neurodevelopmental disorder with hypotonia and cerebellar atrophy, with or without seizures | NEDHCAS; MIM:618879 |
| Q96S52 | PIGS | GPI transamidase component PIG-S | 5 | 0.49 | 0.002 | Glycosylphosphatidylinositol biosynthesis defect 18 | GPIBD18; MIM:618143 |
| P04792 | HSPB1 | Heat shock protein β-1 | 13 | 0.49 | 0.050 | Charcot-Marie-Tooth disease 2F & Neuronopathy, distal hereditary motor, 2B | CMT2F; MIM:606595 & HMN2B; MIM:608634 |
| P21964 | COMT | Catechol O-methyltransferase | 3 | 0.46 | 0.032 | Schizophrenia | SCZD; MIM:181500 |
| Q16643 | DBN1 | Drebrin | 9 | 0.42 | 0.000 | Alzheimer disease | AD; MIM:104300 |
| Q92597 | NDRG1 | Protein NDRG1 | 2 | 0.31 | 0.004 | Charcot-Marie-Tooth disease 4D | CMT4D; MIM:601455 |
| Q6DN90 | IQSEC1 | IQ motif and SEC7 domain-containing protein 1 | 3 | 0.23 | 0.000 | Intellectual developmental disorder with short stature and behavioral abnormalities | IDDSSBA; MIM:618687 |
| Q7L3T8 | PARS2 | Probable proline-tRNA ligase, mitochondrial | 2 | 0.19 | 0.010 | Developmental and epileptic encephalopathy 75 | DEE75; MIM:618437 |
| HLA-DRA | HLA-DPB1 | HLA-DOB | HLA-DRB | TGFB | STAT1 | STAT2 | IL1B | MRC1 (CD206) |
|---|---|---|---|---|---|---|---|---|
| 254× | 183× | n.d. | n.d. | 28× | 7× | 29× | 27× | 3× |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kölbel, H.; Kraft, F.; Hentschel, A.; Czech, A.; Gangfuss, A.; Mohassel, P.; Nguyen, C.; Stenzel, W.; Schara-Schmidt, U.; Preuße, C.; et al. New Insights into the Neuromyogenic Spectrum of a Gain of Function Mutation in SPTLC1. Genes 2022, 13, 893. https://doi.org/10.3390/genes13050893
Kölbel H, Kraft F, Hentschel A, Czech A, Gangfuss A, Mohassel P, Nguyen C, Stenzel W, Schara-Schmidt U, Preuße C, et al. New Insights into the Neuromyogenic Spectrum of a Gain of Function Mutation in SPTLC1. Genes. 2022; 13(5):893. https://doi.org/10.3390/genes13050893
Chicago/Turabian StyleKölbel, Heike, Florian Kraft, Andreas Hentschel, Artur Czech, Andrea Gangfuss, Payam Mohassel, Chi Nguyen, Werner Stenzel, Ulrike Schara-Schmidt, Corinna Preuße, and et al. 2022. "New Insights into the Neuromyogenic Spectrum of a Gain of Function Mutation in SPTLC1" Genes 13, no. 5: 893. https://doi.org/10.3390/genes13050893
APA StyleKölbel, H., Kraft, F., Hentschel, A., Czech, A., Gangfuss, A., Mohassel, P., Nguyen, C., Stenzel, W., Schara-Schmidt, U., Preuße, C., & Roos, A. (2022). New Insights into the Neuromyogenic Spectrum of a Gain of Function Mutation in SPTLC1. Genes, 13(5), 893. https://doi.org/10.3390/genes13050893