
Characterization of Immune-Based Molecular Subtypes and Prognostic Model in Prostate Adenocarcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and methods
2.1. Data Resource
2.2. Characterization of Immune-Based Subtypes
2.3. Evaluation of Immune Characteristics in Subtypes
2.4. Functional Enrichment Analysis
2.5. The Potential Differences of Immune and Chemical Response Prediction
2.6. Analysis of Differentially Expressed Immune-Related Genes
2.7. Survival Analysis and Cox Regression Analysis
2.8. Statistical Analysis
3. Results
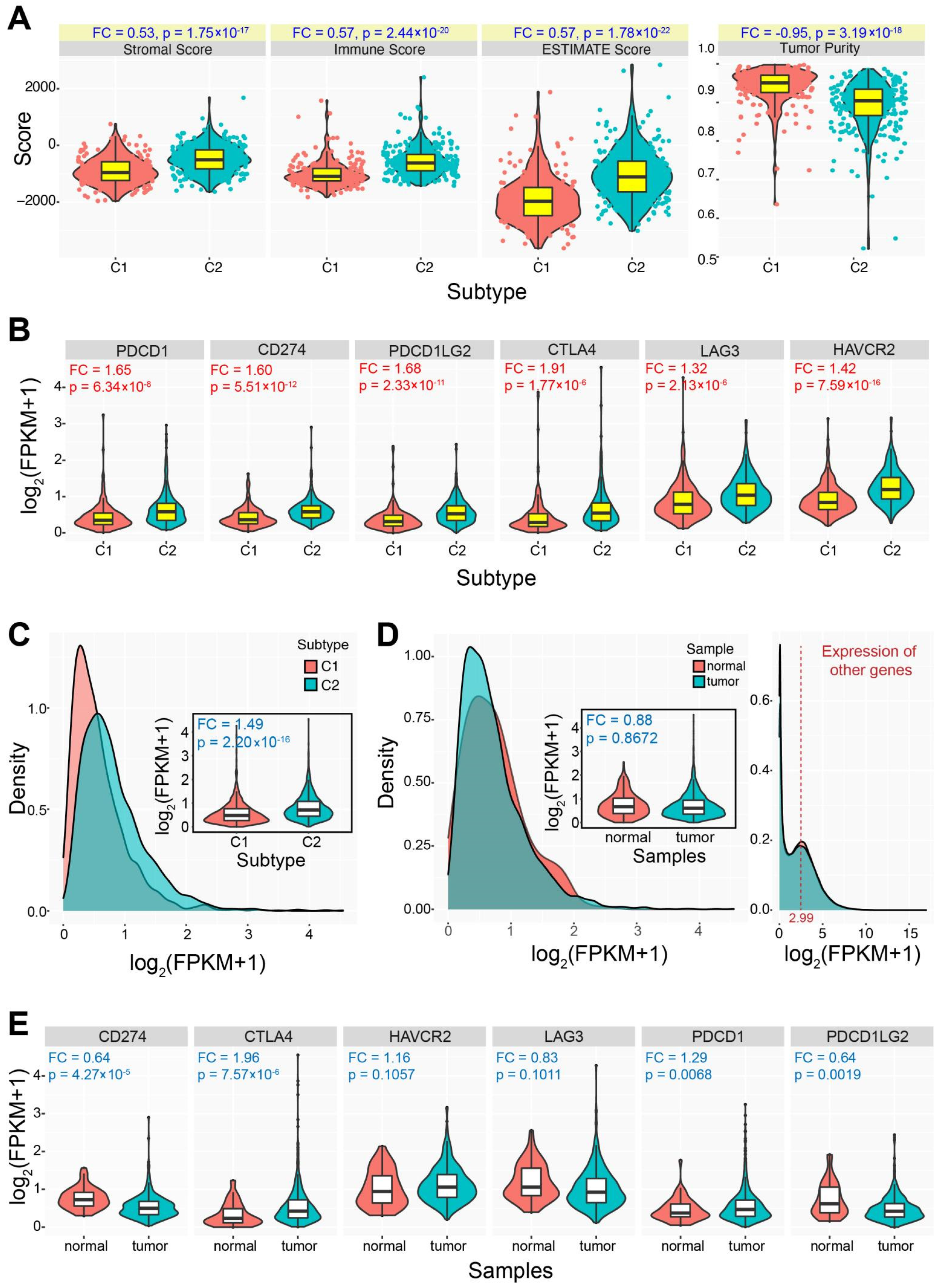
3.1. Two Molecular Subtypes Identified using Immune-Based Genes
3.2. Immune Characteristics of the Two Identified Subtypes
3.3. Differences in Drug Sensitivity between the Two Subtypes
3.4. Difference of Cancer Prognosis between the Two Subtypes
3.5. Construction of Immune-Related Prognostic Model
3.6. The Two Immune-Based Subtypes Show Potential Application in Other Cancers
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation Lists of Involved Cancers in TCGA
References
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Shin, H.-R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef]
- Barbosa, F.G.; Queiroz, M.A.; Nunes, R.F.; Viana, P.C.C.; Marin, J.F.G.; Cerri, G.G.; Buchpiguel, C.A. Revisiting Prostate Cancer Recurrence with PSMA PET: Atlas of Typical and Atypical Patterns of Spread. RadioGraphics 2019, 39, 186–212. [Google Scholar] [CrossRef] [PubMed]
- Gandaglia, G.; Abdollah, F.; Schiffmann, J.; Trudeau, V.; Shariat, S.F.; Kim, S.P.; Perrotte, P.; Montorsi, F.; Briganti, A.; Trinh, Q.-D.; et al. Distribution of metastatic sites in patients with prostate cancer: A population-based analysis. Prostate 2013, 74, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, G.; Buonerba, C.; Kantoff, P.W. Immunotherapy for the treatment of prostate cancer. Nat. Rev. Clin. Oncol. 2011, 8, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Immune suppression in cancer: Effects on immune cells, mechanisms and future therapeutic intervention. Semin. Cancer Biol. 2006, 16, 3–15. [Google Scholar] [CrossRef]
- Hossain, K.; Nahar, K.; Donkor, O.; Apostolopoulos, V. Immune-based therapies for metastatic prostate cancer: An update. Immunotherapy 2018, 10, 283–298. [Google Scholar] [CrossRef]
- McNeel, D.G.; Malkovsky, M. Immune-based therapies for prostate cancer. Immunol. Lett. 2005, 96, 3–9. [Google Scholar] [CrossRef]
- Koźmiński, P.; Halik, P.K.; Chesori, R.; Gniazdowska, E. Overview of Dual-Acting Drug Methotrexate in Different Neurological Diseases, Autoimmune Pathologies and Cancers. Int. J. Mol. Sci. 2020, 21, 3483. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Fidelito, G.; Watt, M.J.; Taylor, R.A. Personalized Medicine for Prostate Cancer: Is Targeting Metabolism a Reality? Front. Oncol. 2021, 11, 778761. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.; Li, J.; Su, Q.; Ma, H.; Wei, Q.; Li, H.; Gao, G. rAAV-delivered PTEN therapeutics for prostate cancer. Mol. Ther. Nucleic. Acids. 2022, 27, 122–132. [Google Scholar] [CrossRef] [PubMed]
- De Moraes, R.P.; Pimenta, R.; Mori, F.N.C.; Dos Santos, G.A.; Viana, N.I.; Guimarães, V.R.; De Camargo, J.A.; Leite, K.R.M.; Srougi, M.; Nahas, W.C.; et al. Tissue expression of MMP-9, TIMP-1, RECK, and miR338-3p in prostate gland: Can it predict cancer? Mol. Biol. Res. Commun. 2021, 10, 149–156. [Google Scholar]
- Kan, Y.; Li, B.; Yang, D.; Liu, Y.; Liu, J.; Yang, C.; Mao, L. Emerging Roles of Long Non-coding RNAs as Novel Biomarkers in the Diagnosis and Prognosis of Prostate Cancer. Discov. Med. 2021, 32, 29–37. [Google Scholar]
- Singh, N.; Ramnarine, V.R.; Song, J.H.; Pandey, R.; Padi, S.K.R.; Nouri, M.; Olive, V.; Kobelev, M.; Okumura, K.; McCarthy, D.; et al. The long noncoding RNA H19 regulates tumor plasticity in neuroendocrine prostate cancer. Nat. Commun. 2021, 12, 7349. [Google Scholar] [CrossRef]
- Meng, J.; Lu, X.; Jin, C.; Zhou, Y.; Ge, Q.; Zhou, J.; Hao, Z.; Yan, F.; Zhang, M.; Liang, C. Integrated multi-omics data reveals the molecular subtypes and guides the androgen receptor signalling inhibitor treatment of prostate cancer. Clin. Transl. Med. 2021, 11, e655. [Google Scholar] [CrossRef]
- Goldman, M.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. biorxiv 2019, 326470. [Google Scholar] [CrossRef] [Green Version]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Andorf, S.; Gomes, L.; Dunn, P.; Schaefer, H.; Pontius, J.; Berger, P.; Desborough, V.; Smith, T.; Campbell, J.; et al. ImmPort: Disseminating data to the public for the future of immunology. Immunol. Res. 2014, 58, 234–239. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Dunn, P.; Thomas, C.; Smith, B.; Schaefer, H.; Chen, J.; Hu, Z.; Zalocusky, K.A.; Shankar, R.D.; Shen-Orr, S.S.; et al. ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci. Data 2018, 5, 180015. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeser, D.; Gruener, R.F.; Huang, R.S. oncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief. Bioinform. 2021, 22, bbab260. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Belinda, P.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Kishore, J.; Goel, M.; Khanna, P. Understanding survival analysis: Kaplan-Meier estimate. Int. J. Ayurveda Res. 2010, 1, 274–278. [Google Scholar] [CrossRef] [Green Version]
- Robles, A.; Harris, C.C. Clinical Outcomes and Correlates of TP53 Mutations and Cancer. Cold Spring Harb. Perspect. Biol. 2009, 2, a001016. [Google Scholar] [CrossRef] [Green Version]
- Akinyemiju, T.; Oyekunle, T.; Salako, O.; Gupta, A.; Alatise, O.; Ogun, G.; Adeniyi, A.; Deveaux, A.; Hall, A.; Ayandipo, O.; et al. Metabolic Syndrome and Risk of Breast Cancer by Molecular Subtype: Analysis of the MEND Study. Clin. Breast Cancer 2022, 22, e463–e472. [Google Scholar] [CrossRef] [PubMed]
- Sengoz, T.; Karakaya, Y.A.; Gultekin, A.; Yaylali, O.; Senol, H.; Yuksel, D. Relationships of 18F-FDG uptake by primary tumors with prognostic factors and molecular subtype in ductal breast cancer. Rev. Española De Med. Nucl. E Imagen Mol. (Engl. Ed. ) 2021, 41, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Wang, J.; Zhang, M.; Wang, S.; Zhao, L.; Yang, H.; Wu, J.; Cui, B. Comprehensive Analysis of Subtype-Specific Molecular Characteristics of Colon Cancer: Specific Genes, Driver Genes, Signaling Pathways, and Immunotherapy Responses. Front. Cell Dev. Biol. 2021, 9, 758776. [Google Scholar] [CrossRef] [PubMed]
- Vuaroqueaux, V.; Musch, A.; Kobelt, D.; Risch, T.; Herrmann, P.; Burock, S.; Peille, A.-L.; Yaspo, M.-L.; Fiebig, H.-H.; Stein, U. Elevated MACC1 Expression in Colorectal Cancer Is Driven by Chromosomal Instability and Is Associated with Molecular Subtype and Worse Patient Survival. Cancers 2022, 14, 1749. [Google Scholar] [CrossRef]
- Yu, X.; Yu, B.; Fang, W.; Xiong, J.; Ma, M. Identification hub genes of consensus molecular subtype correlation with immune infiltration and predict prognosis in gastric cancer. Discov Oncol. 2021, 12, 41. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar]
- Fontugne, J.; Cai, P.Y.; Alnajar, H.; Bhinder, B.; Park, K.; Ye, H.; Beg, S.; Sailer, V.; Siddiqui, J.; Blattner-Johnson, M.; et al. Collision tumors revealed by prospectively assessing subtype-defining molecular alterations in 904 individual prostate cancer foci. JCI Insight 2022, 7, e155309. [Google Scholar] [CrossRef]
- Kobelyatskaya, A.; Pudova, E.; Snezhkina, A.; Fedorova, M.; Pavlov, V.; Guvatova, Z.; Savvateeva, M.; Melnikova, N.; Dmitriev, A.; Trofimov, D.; et al. Impact TMPRSS2–ERG Molecular Subtype on Prostate Cancer Recurrence. Life 2021, 11, 588. [Google Scholar] [CrossRef]
- Han, H.; Lee, H.H.; Choi, K.; Moon, Y.J.; Heo, J.E.; Ham, W.S.; Jang, W.S.; Rha, K.H.; Cho, N.H.; Giancotti, F.G.; et al. Prostate epithelial genes define therapy-relevant prostate cancer molecular subtype. Prostate Cancer Prostatic Dis. 2021, 24, 1080–1092. [Google Scholar] [CrossRef]
- Yeku, O.; Slovin, S.F. Immune Therapy for Prostate Cancer. Cancer J. 2016, 22, 334–341. [Google Scholar] [CrossRef]
- Claps, M.; Mennitto, A.; Guadalupi, V.; Sepe, P.; Stellato, M.; Zattarin, E.; Gillessen, S.S.; Sternberg, C.N.; Berruti, A.; De Braud, F.G.M.; et al. Immune-checkpoint inhibitors and metastatic prostate cancer therapy: Learning by making mistakes. Cancer Treat. Rev. 2020, 88, 102057. [Google Scholar] [CrossRef] [PubMed]
- Ngan, H.-L.; Law, C.-H.; Choi, Y.C.Y.; Chan, J.Y.-S.; Lui, V.W.Y. Precision drugging of the MAPK pathway in head and neck cancer. Npj Genom. Med. 2022, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Liu, R.; Hu, X.; Liang, T.; Zhou, Z.; Huang, Z. MAPK signaling pathway-targeted marine compounds in cancer therapy. J. Cancer Res. Clin. Oncol. 2021, 147, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Banerjee, S.; Mondal, A.; Chakraborty, U.; Pumarol, J.; Croley, C.; Bishayee, A. Targeting the JAK/STAT Signaling Pathway Using Phytocompounds for Cancer Prevention and Therapy. Cells 2020, 9, 1451. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Damiano, J.S.; Wasserman, E. Molecular Pathways: Blockade of the PRLR Signaling Pathway as a Novel Antihormonal Approach for the Treatment of Breast and Prostate Cancer. Clin. Cancer Res. 2013, 19, 1644–1650. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, C.C.; Bates, S.E. Targeting Prolactin Receptor (PRLR) Signaling in PRLR-Positive Breast and Prostate Cancer. Oncologist 2016, 21, 523–526. [Google Scholar] [CrossRef]
- Arnold, R.S.; He, J.; Remo, A.; Ritsick, D.; Yin-Goen, Q.; Lambeth, J.D.; Datta, M.W.; Young, A.N.; Petros, J.A. Nox1 Expression Determines Cellular Reactive Oxygen and Modulates c-fos-Induced Growth Factor, Interleukin-8, and Cav-1. Am. J. Pathol. 2007, 171, 2021–2032. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.D.; Sun, C.; Lambeth, J.D.; Marshall, F.; Amin, M.; Chung, L.; Petros, J.A.; Arnold, R.S. Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate 2004, 62, 200–207. [Google Scholar] [CrossRef]
- Chauvin, M.; Garambois, V.; Colombo, P.-E.; Chentouf, M.; Gros, L.; Brouillet, J.-P.; Robert, B.; Jarlier, M.; Dumas, K.; Martineau, P.; et al. Anti-Müllerian hormone (AMH) autocrine signaling promotes survival and proliferation of ovarian cancer cells. Sci. Rep. 2021, 11, 2231. [Google Scholar] [CrossRef]
- Shrikhande, L.; Shrikhande, B.; Shrikhande, A. AMH and Its Clinical Implications. J. Obstet. Gynecol. India 2020, 70, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Ainiwaer, Z.; Maisaidi, R.; Liu, J.; Han, L.; Husaiyin, S.; Niyazi, M. Genetic polymorphisms of PGF and TNFAIP2 genes related to cervical cancer risk among Uygur females from China. BMC Med. Genet. 2020, 21, 212. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, L.; Kang, Y.; Xia, D.; Ren, Y.; Yang, X.; Xiang, Y.; Tang, L.; Ren, D.; Yu, J.; Wang, J.; et al. Characterization of Immune-Based Molecular Subtypes and Prognostic Model in Prostate Adenocarcinoma. Genes 2022, 13, 1087. https://doi.org/10.3390/genes13061087
Guo L, Kang Y, Xia D, Ren Y, Yang X, Xiang Y, Tang L, Ren D, Yu J, Wang J, et al. Characterization of Immune-Based Molecular Subtypes and Prognostic Model in Prostate Adenocarcinoma. Genes. 2022; 13(6):1087. https://doi.org/10.3390/genes13061087
Chicago/Turabian StyleGuo, Li, Yihao Kang, Daoliang Xia, Yujie Ren, Xueni Yang, Yangyang Xiang, Lihua Tang, Dekang Ren, Jiafeng Yu, Jun Wang, and et al. 2022. "Characterization of Immune-Based Molecular Subtypes and Prognostic Model in Prostate Adenocarcinoma" Genes 13, no. 6: 1087. https://doi.org/10.3390/genes13061087