
Identification of COVID-19-Associated DNA Methylation Variations by Integrating Methylation Array and scRNA-Seq Data at Cell-Type Resolution

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
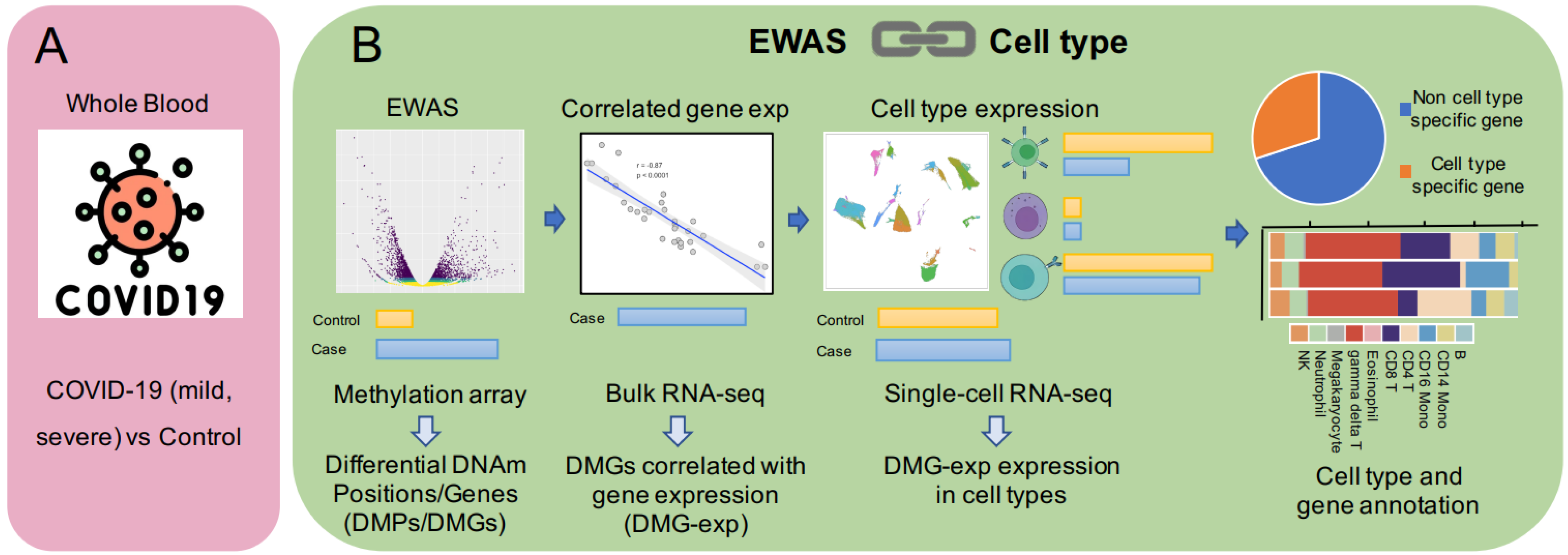
2.1. The Construction of the Pipeline
2.2. Data Collection
2.3. Differential Methylation Analysis
2.4. DNAm-mRNA Correlation Analysis
2.5. Single-Cell RNA-Seq Data Analysis
3. Results
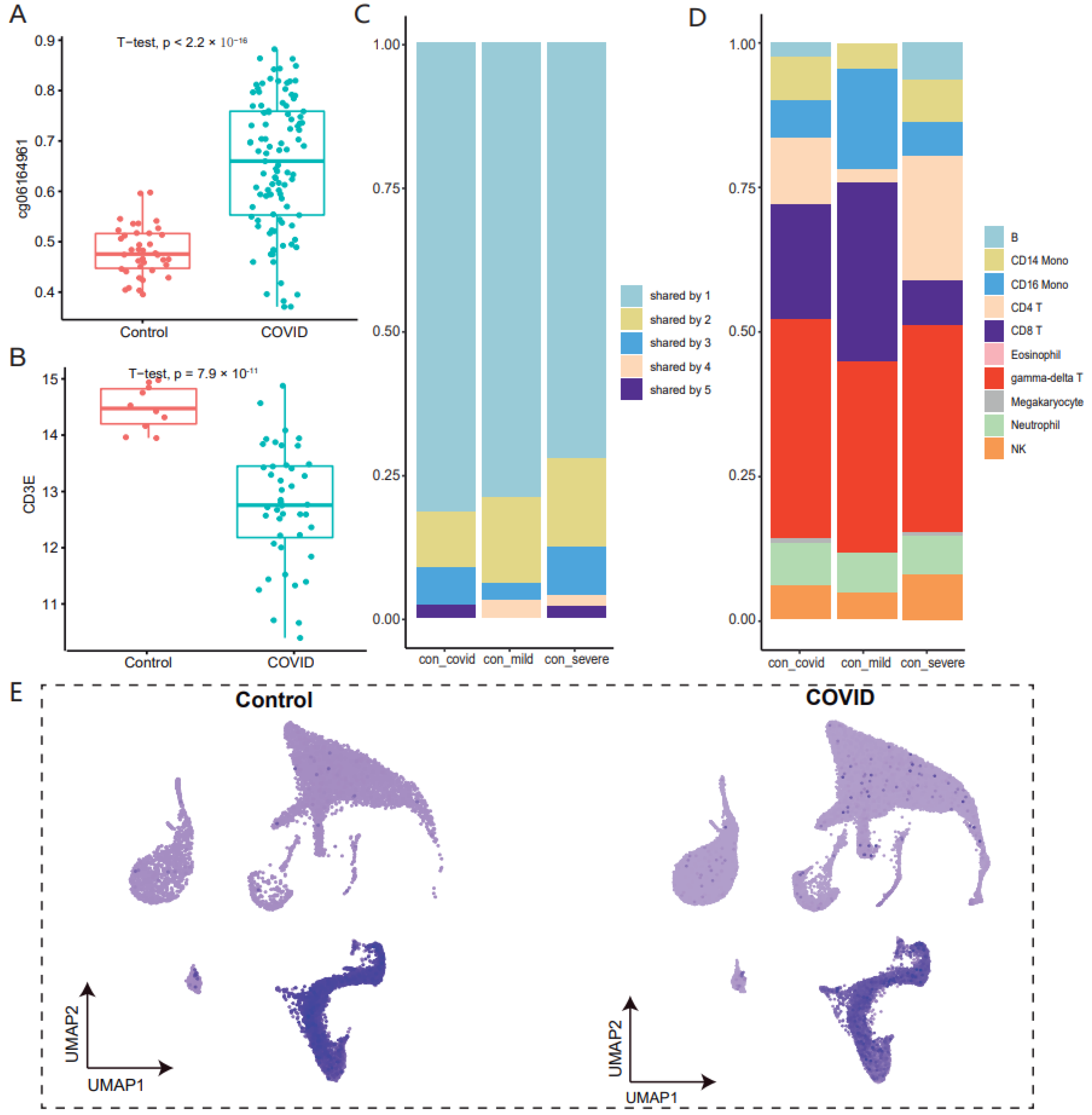
3.1. DNA Methylation Variations in Whole Blood of COVID-19 at the Cell-Type Resolution
3.2. Methylated Variant Genes Associated with COVID-19 Have Significant Cell-Type Specificity
3.3. The Majority of Methylated Variant Genes Are Associated with Gamma-Delta T Cells in COVID-19
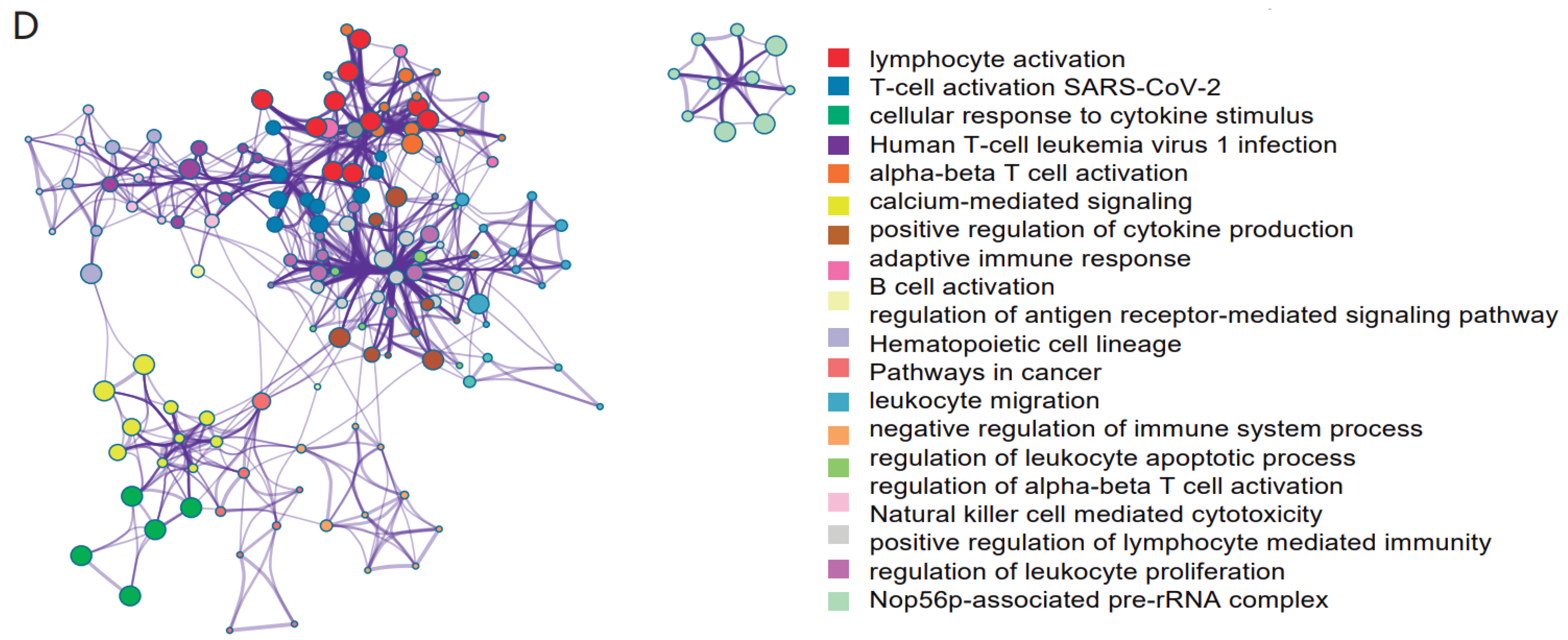
3.4. Multiple Cell-Type-Related Genes May Be Important Regulatory Genes of COVID-19 Immune Response
4. Discussion
5. Conclusions
6. Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Yao, C.; Bora, S.A.; Parimon, T.; Zaman, T.; Friedman, O.A.; Palatinus, J.A.; Surapaneni, N.S.; Matusov, Y.P.; Cerro Chiang, G.; Kassar, A.G.; et al. Cell-Type-Specific Immune Dysregulation in Severely Ill COVID-19 Patients. Cell Rep. 2021, 34, 108590. [Google Scholar] [CrossRef]
- Meckiff, B.J.; Ramirez-Suastegui, C.; Fajardo, V.; Chee, S.J.; Kusnadi, A.; Simon, H.; Eschweiler, S.; Grifoni, A.; Pelosi, E.; Weiskopf, D.; et al. Imbalance of Regulatory and Cytotoxic SARS-CoV-2-Reactive CD4(+) T Cells in COVID-19. Cell 2020, 183, 1340–1353.e1316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Wang, X.M.; Xing, X.; Xu, Z.; Zhang, C.; Song, J.W.; Fan, X.; Xia, P.; Fu, J.L.; Wang, S.Y.; et al. Single-cell landscape of immunological responses in patients with COVID-19. Nat. Immunol. 2020, 21, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Corley, M.J.; Pang, A.P.S.; Dody, K.; Mudd, P.A.; Patterson, B.K.; Seethamraju, H.; Bram, Y.; Peluso, M.J.; Torres, L.; Iyer, N.S.; et al. Genome-wide DNA methylation profiling of peripheral blood reveals an epigenetic signature associated with severe COVID-19. J. Leukoc. Biol. 2021, 110, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Wakim, L.M.; Gupta, N.; Mintern, J.D.; Villadangos, J.A. Enhanced survival of lung tissue-resident memory CD8(+) T cells during infection with influenza virus due to selective expression of IFITM3. Nat. Immunol. 2013, 14, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Barwick, B.G.; Scharer, C.D.; Martinez, R.J.; Price, M.J.; Wein, A.N.; Haines, R.R.; Bally, A.P.R.; Kohlmeier, J.E.; Boss, J.M. B cell activation and plasma cell differentiation are inhibited by de novo DNA methylation. Nat. Commun. 2018, 9, 1900. [Google Scholar] [CrossRef] [Green Version]
- Bernardes, J.P.; Mishra, N.; Tran, F.; Bahmer, T.; Best, L.; Blase, J.I.; Bordoni, D.; Franzenburg, J.; Geisen, U.; Josephs-Spaulding, J.; et al. Longitudinal Multi-omics Analyses Identify Responses of Megakaryocytes, Erythroid Cells, and Plasmablasts as Hallmarks of Severe COVID-19. Immunity 2020, 53, 1296–1314.e1299. [Google Scholar] [CrossRef]
- Balnis, J.; Madrid, A.; Hogan, K.J.; Drake, L.A.; Chieng, H.C.; Tiwari, A.; Vincent, C.E.; Chopra, A.; Vincent, P.A.; Robek, M.D.; et al. Blood DNA methylation and COVID-19 outcomes. Clin. Epigen. 2021, 13, 118. [Google Scholar] [CrossRef]
- Carter, B.; Zhao, K. The epigenetic basis of cellular heterogeneity. Nat. Rev. Genet. 2021, 22, 235–250. [Google Scholar] [CrossRef]
- Lee, J.; Hyeon, D.Y.; Hwang, D. Single-cell multiomics: Technologies and data analysis methods. Exp. Mol. Med. 2020, 52, 1428–1442. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Liu, J.; Beck, S.; Pan, S.; Capper, D.; Lechner, M.; Thirlwell, C.; Breeze, C.E.; Teschendorff, A.E. A pan-tissue DNA methylation atlas enables in silico decomposition of human tissue methylomes at cell-type resolution. Nat. Methods 2022, 19, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.; Kang, H.; Xiong, Z.; Ma, Y.; Jin, T.; Gong, Z.; Yi, L.; Zhang, M.; Wu, S.; Wang, G.; et al. scMethBank: A database for single-cell whole genome DNA methylation maps. Nucleic Acids Res. 2022, 50, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Liang, F.; Li, M.; Zou, D.; Sun, S.; Zhao, Y.; Zhao, W.; Bao, Y.; Xiao, J.; Zhang, Z. MethBank 3.0: A database of DNA methylomes across a variety of species. Nucleic Acids Res. 2018, 46, D288–D295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Zou, D.; Li, Z.; Gao, R.; Sang, J.; Zhang, Y.; Li, R.; Xia, L.; Zhang, T.; Niu, G.; et al. EWAS Atlas: A curated knowledgebase of epigenome-wide association studies. Nucleic Acids Res. 2019, 47, D983–D988. [Google Scholar] [CrossRef]
- Xiong, Z.; Li, M.; Yang, F.; Ma, Y.; Sang, J.; Li, R.; Li, Z.; Zhang, Z.; Bao, Y. EWAS Data Hub: A resource of DNA methylation array data and metadata. Nucleic Acids Res. 2020, 48, D890–D895. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Z.; Yang, F.; Li, M.; Ma, Y.; Zhao, W.; Wang, G.; Li, Z.; Zheng, X.; Zou, D.; Zong, W.; et al. EWAS Open Platform: Integrated data, knowledge and toolkit for epigenome-wide association study. Nucleic Acids Res. 2022, 50, D1004–D1009. [Google Scholar] [CrossRef]
- Hannon, E.; Dempster, E.L.; Mansell, G.; Burrage, J.; Bass, N.; Bohlken, M.M.; Corvin, A.; Curtis, C.J.; Dempster, D.; Di Forti, M.; et al. DNA methylation meta-analysis reveals cellular alterations in psychosis and markers of treatment-resistant schizophrenia. eLife 2021, 10, e58430. [Google Scholar] [CrossRef]
- Jia, T.; Chu, C.; Liu, Y.; van Dongen, J.; Papastergios, E.; Armstrong, N.J.; Bastin, M.E.; Carrillo-Roa, T.; den Braber, A.; Harris, M.; et al. Epigenome-wide meta-analysis of blood DNA methylation and its association with subcortical volumes: Findings from the ENIGMA Epigenetics Working Group. Mol. Psychiatry 2021, 26, 3884–3895. [Google Scholar] [CrossRef] [Green Version]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinho, R.M.; Maga, E.A. DNA methylation as a regulator of intestinal gene expression. Br. J. Nutr. 2021, 126, 1611–1625. [Google Scholar] [CrossRef]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Nabais, M.F.; Laws, S.M.; Lin, T.; Vallerga, C.L.; Armstrong, N.J.; Blair, I.P.; Kwok, J.B.; Mather, K.A.; Mellick, G.D.; Sachdev, P.S.; et al. Meta-analysis of genome-wide DNA methylation identifies shared associations across neurodegenerative disorders. Genome. Biol. 2021, 22, 90. [Google Scholar] [CrossRef] [PubMed]
- Overmyer, K.A.; Shishkova, E.; Miller, I.J.; Balnis, J.; Bernstein, M.N.; Peters-Clarke, T.M.; Meyer, J.G.; Quan, Q.; Muehlbauer, L.K.; Trujillo, E.A.; et al. Large-Scale Multi-omic Analysis of COVID-19 Severity. Cell Syst. 2021, 12, 23–40.e27. [Google Scholar] [CrossRef]
- Bost, P.; De Sanctis, F.; Cane, S.; Ugel, S.; Donadello, K.; Castellucci, M.; Eyal, D.; Fiore, A.; Anselmi, C.; Barouni, R.M.; et al. Deciphering the state of immune silence in fatal COVID-19 patients. Nat. Commun. 2021, 12, 1428. [Google Scholar] [CrossRef]
- Schulte-Schrepping, J.; Reusch, N.; Paclik, D.; Bassler, K.; Schlickeiser, S.; Zhang, B.; Kramer, B.; Krammer, T.; Brumhard, S.; Bonaguro, L.; et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 2020, 182, 1419–1440.e1423. [Google Scholar] [CrossRef]
- Xiong, Z.; Li, M.; Ma, Y.; Li, R.; Bao, Y. GMQN: A Reference-Based Method for Correcting Batch Effects and Probe Bias in HumanMethylation BeadChip. Front Genet. 2021, 12, 810985. [Google Scholar] [CrossRef]
- Troyanskaya, O.; Cantor, M.; Sherlock, G.; Brown, P.; Hastie, T.; Tibshirani, R.; Botstein, D.; Altman, R.B. Missing value estimation methods for DNA microarrays. Bioinformatics 2001, 17, 520–525. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Morris, T.J.; Webster, A.P.; Yang, Z.; Beck, S.; Feber, A.; Teschendorff, A.E. ChAMP: Updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 2017, 33, 3982–3984. [Google Scholar] [CrossRef] [Green Version]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e1821. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, X.; Ang, K.S.; Ling, J.; Sethi, R.; Lee, N.Y.S.; Ginhoux, F.; Chen, J. DISCO: A database of Deeply Integrated human Single-Cell Omics data. Nucleic Acids Res. 2022, 50, D596–D602. [Google Scholar] [CrossRef] [PubMed]
- Ribot, J.C.; Lopes, N. Silva-Santos B: Gammadelta T cells in tissue physiology and surveillance. Nat. Rev. Immunol. 2021, 21, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.; Malkovsky, M.; Carding, S.R. Gamma/delta T lymphocytes in viral infections. J. Leukoc. Biol. 1995, 58, 277–283. [Google Scholar] [CrossRef]
- Miyashita, M.; Shimizu, T.; Ashihara, E.; Ukimura, O. Strategies to Improve the Antitumor Effect of gammadelta T Cell Immunotherapy for Clinical Application. Int. J. Mol. Sci. 2021, 22, 8910. [Google Scholar] [CrossRef]
- Poccia, F.; Agrati, C.; Castilletti, C.; Bordi, L.; Gioia, C.; Horejsh, D.; Ippolito, G.; Chan, P.K.; Hui, D.S.; Sung, J.J.; et al. Anti-severe acute respiratory syndrome coronavirus immune responses: The role played by V gamma 9V delta 2 T cells. J. Infect. Dis. 2006, 193, 1244–1249. [Google Scholar] [CrossRef]
- von Massow, G.; Oh, S.; Lam, A.; Gustafsson, K. Gamma Delta T Cells and Their Involvement in COVID-19 Virus Infections. Front. Immunol. 2021, 12, 741218. [Google Scholar] [CrossRef]
- Shi, H.; Zuo, Y.; Yalavarthi, S.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Woodward, W.; Lezak, S.P.; Lugogo, N.L.; et al. Neutrophil calprotectin identifies severe pulmonary disease in COVID-19. J. Leukoc. Biol. 2021, 109, 67–72. [Google Scholar] [CrossRef]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in Inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef]
- Mahler, M.; Meroni, P.L.; Infantino, M.; Buhler, K.A.; Fritzler, M.J. Circulating Calprotectin as a Biomarker of COVID-19 Severity. Expert. Rev. Clin. Immunol. 2021, 17, 431–443. [Google Scholar] [CrossRef]
- Imai, T.; Yasuda, N. Therapeutic intervention of inflammatory/immune diseases by inhibition of the fractalkine (CX3CL1)-CX3CR1 pathway. Inflamm. Regen. 2016, 36, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazolo, T.; Lima, K.; Fontoura, J.C.; de Souza, P.O.; Hilario, G.; Zorzetto, R.; Junior, L.R.; Pscheidt, V.M.; de Castilhos Ferreira Neto, J.; Haubert, A.F.; et al. Pediatric COVID-19 patients in South Brazil show abundant viral mRNA and strong specific anti-viral responses. Nat. Commun. 2021, 12, 6844. [Google Scholar] [CrossRef] [PubMed]
- Sundararaj, S.; Ravindran, A.; Casarotto, M.G. AHNAK: The quiet giant in calcium homeostasis. Cell Calcium. 2021, 96, 102403. [Google Scholar] [CrossRef] [PubMed]
- Haas, P. Systematic Investigation of Host-Pathogen Interactions Identifies Novel Drug Targets for HIV and SARS-CoV-2. 2021. Available online: https://escholarship.org/uc/item/84x302rk (accessed on 1 January 2021).
- Meng, H.; Long, Q.; Wang, R.; Zhou, X.; Su, H.; Wang, T.; Li, Y. Identification of the Key Immune-Related Genes in Chronic Obstructive Pulmonary Disease Based on Immune Infiltration Analysis. Int. J. Chron. Obstruct. Pulmon. Dis. 2022, 17, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Wang, K.; Gu, Y.; Chen, Y.; Nan, X.; Xing, J.; Cui, Q.; Chen, Y.; Ge, Q.; Zhao, H. TRAF3IP3, a novel autophagy up-regulated gene, is involved in marginal zone B lymphocyte development and survival. Clin. Exp. Immunol. 2015, 182, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Li, J.; Zhang, R.; Cai, Y.; Wang, C.; Qi, S.; Chen, S.; Liang, X.; Qi, N.; Hou, F. TRAF3IP3 mediates the recruitment of TRAF3 to MAVS for antiviral innate immunity. EMBO J. 2019, 38, e102075. [Google Scholar] [CrossRef]
- Williams, D.R.; Shifley, E.T.; Braunreiter, K.M.; Cole, S.E. Disruption of somitogenesis by a novel dominant allele of Lfng suggests important roles for protein processing and secretion. Development 2016, 143, 822–830. [Google Scholar]
- Breikaa, R.M.; Lilly, B. The Notch Pathway: A Link Between COVID-19 Pathophysiology and Its Cardiovascular Complications. Front. Cardiovasc. Med. 2021, 8, 681948. [Google Scholar] [CrossRef]
- Mittelstaedt, N.N.; Becker, A.L.; de Freitas, D.N.; Zanin, R.F.; Stein, R.T.; Duarte de Souza, A.P. DNA Methylation and Immune Memory Response. Cells 2021, 10, 2943. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Samples | Type | Platform | Annotation |
|---|---|---|---|---|
| GSE153712 | 38 | Control | EPIC | Bulk-tissue |
| GSE174818 | 101 | COVID-19 | EPIC | Bulk-tissue |
| GSE157103 | 101 | COVID-19 | RNA-seq | Bulk-tissue |
| GSE157344 | 31 | Control; Mild; Severe | 10× scRNA-seq | Single-cell |
| EGAS0000001004571 | 16 | Control; Mild; Severe | 10× scRNA-seq | Single-cell |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, G.; Xiong, Z.; Yang, F.; Zheng, X.; Zong, W.; Li, R.; Bao, Y. Identification of COVID-19-Associated DNA Methylation Variations by Integrating Methylation Array and scRNA-Seq Data at Cell-Type Resolution. Genes 2022, 13, 1109. https://doi.org/10.3390/genes13071109
Wang G, Xiong Z, Yang F, Zheng X, Zong W, Li R, Bao Y. Identification of COVID-19-Associated DNA Methylation Variations by Integrating Methylation Array and scRNA-Seq Data at Cell-Type Resolution. Genes. 2022; 13(7):1109. https://doi.org/10.3390/genes13071109
Chicago/Turabian StyleWang, Guoliang, Zhuang Xiong, Fei Yang, Xinchang Zheng, Wenting Zong, Rujiao Li, and Yiming Bao. 2022. "Identification of COVID-19-Associated DNA Methylation Variations by Integrating Methylation Array and scRNA-Seq Data at Cell-Type Resolution" Genes 13, no. 7: 1109. https://doi.org/10.3390/genes13071109