
Genome Sequencing of Consanguineous Family Implicates Ubiquitin-Specific Protease 53 (USP53) Variant in Psychosis/Schizophrenia: Wild-Type Expression in Murine Hippocampal CA 1–3 and Granular Dentate with AMPA Synapse Interactions
 , and
, and
Abstract
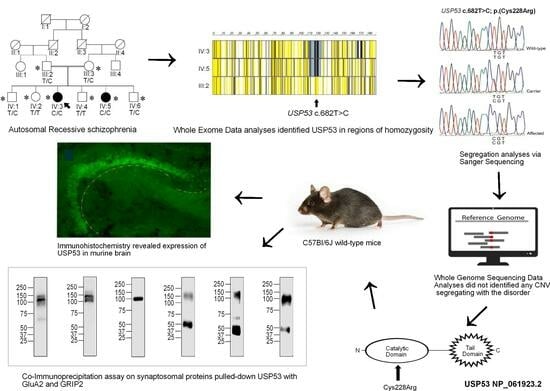
:
1. Introduction
2. Methods
2.1. Subjects
2.2. Genetics: Sequencing and Analysis
2.3. Structural Models
2.4. Immunohistochemistry
2.5. Immunoprecipitation and Western Blots
3. Results
3.1. Pedigree PSY01
3.2. Case Summaries
3.2.1. Proband IV:3
3.2.2. Proband IV:5
3.3. Variant Identification
3.3.1. Exome Sequencing Identifies USP53 as a Sole VOI
3.3.2. Genome Sequencing Did Not Yield Additional Pathogenic SNVs
3.3.3. Genome Sequencing Did Not Yield a Likely Pathogenic CNV
3.3.4. Computational Modeling of USP53 Variants
3.3.5. USP53 Is Expressed in the Mouse Hippocampus
3.3.6. Anti-USP53 Pulls-Down GRIP2 and GRIA2 (GluA2) from Murine Synaptosomes
4. Discussion
4.1. USP53 Structure and Function
4.2. Murine USP53 Cys228Ser (mambo) Variant
4.3. Human USP53 Pathogenic Variants
4.4. Ubiquitin, USP53 Interactome, and the AMPAR Synapse

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ethnicity/ Origin | Onset Age ‡ | Gender | Consanguinity | Variant | USP53 Transcript | Zygosity | Variant Type | Phenotype ± Comorbidities | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Pakistani | 16 years | F | Yes | c.682T>C; p.Cys228Arg | NM_019050.2 | Homozygous | Missense | Psychosis w/o cholestasis | Current |
| Syrian | 4 months | F | Yes | c.238-1G>C | NM_019050.2 | Homozygous | Splice site | Cholestasis | [55] |
| Brazilian | 10 days | M | No | c.1687_1688delinsC p.Ser563Profs * 25 | NM_019050.2 | Homozygous | Frameshift | Cholestasis + Jaundice | [56] |
| Saudi Arabian | N/A | F | Yes | c.951delT p.[Phe317Leufs*6] | NM_019050.2 | Homozygous | Frameshift | Hypocalcemia itching, deafness w/o cholestasis | [57] |
| Ashkenazi | 15 years‡ | F | NA * | c.1295_1299delTAAGT p.Leu432fs | NA * | Heterozygous | Frameshift | Bipolar I | [41] |
| Chinese | 3 days | F | No | c.1012C>T p.Arg338Ter | NM_019050.2 | Homozygous | Nonsense | Jaundice | [58] |
| Chinese | 2 days | M | No | c.169C>T/c.831_832insAG p.Arg57Ter/p.Val279GlufsTer16 | NM_019050.2 | Compound heterozygous | Frameshift | Jaundice | [58] |
| Chinese | 6 months | F | No | c.569+2T>C/c.878G>T p:?/p.Gly293Val | NM_019050.2 | Compound heterozygous | Splice/Mis-sense | Jaundice | [58] |
| Chinese | 5 months | M | No | c.581delA/c.1012C>T p.Arg195GlufsTer38/p.Arg338Ter | NM_019050.2 | Compound heterozygous | Frameshift/Nonsense | Jaundice | [58] |
| Chinese | 1 month | F | No | c.1012C>T/c.1426C>T p.Arg338Ter/p.Arg476Ter | NM_019050.2 | Compound heterozygous | Nonsense | Jaundice | [58] |
| Chinese | 5 months | M | No | c.395A>G/c.1558C>T p.His132Arg/p.Arg520Ter | NM_019050.2 | Compound heterozygous | Missense/Nonsense | Jaundice + hearing loss | [58] |
| Chinese | 7 months | M | No | c.297G>Tc.1012C>T p.Arg99Ser/p.Arg338Ter | NM_019050.2 | Compound heterozygous | Missense/Nonsense | Jaundice | [58] |
| Turkish | 3 months | F | No | Deletion of 1st coding exon | NM_01050.2 | Homozygous | Gross del | Cholestasis | [59] |
| Turkish | 2 months | M | No | Deletion of 1st coding exon | NM_019050.2 | Homozygous | Gross del | Cholestasis with heart failure | [59] |
| Middle Eastern | 5 months | F | No | c.145–11_167del | NM_019050.2 | Homozygous | Gross del | Cholestasis | [59] |
| Middle Eastern | 7 years | M | No | c.145–11_167del | NM_019050.2 | Homozygous | Gross del | Cholestasis | [59] |
| North African | 15 years | M | No | c.725C>T p.Pro242Leu | NM_019050.2 | Homozygous | Missense | Cholestasis | [59] |
| South Asian | 4 years | M | No | c.510delA p.Ser171ArgfsTer62 | NM_019050.2 | Homozygous | Frameshift | Cholestasis | [59] |
| Japanese | 16 years | F | Yes | c.1744C>T p. Arg582 * | NM_019050.2 | Homozygous | Nonsense | Benign recurrent intrahepatic cholestasis + itching + hypothyroid | [39] |
| Dagestan/ Russian | Neonatal | M | Yes | c.1017_1057del p.Cys339Trpfs | NM_019050.2 | Homozygous | Frameshift | Prolonged jaundice | [60] |
| Indian | 6 months | M | Yes | c.822+1delG | NM_019050.2 | Homozygous | Splice site | Jaundice with pruritus | [61] |
| Pakistani | 8 months | M | Yes | c.1524T>G p.Tyr508 * | NM_019050.2 | Homozygous | Nonsense | Jaundice + thrombocytosis+ itching + pigmented stools | [62] |
| Pakistani | NA * | NA * | NA * | c.169C>T p.Arg57 * | NM_019050.2 | Homozygous | Nonsense | Early-onset intrahepatic cholestasis | [62] |
| Pakistani | NA * | NA * | NA * | c.475_476delCT p.Leu159fs | NM_019050.2 | Homozygous | Frameshift | Early-onset intrahepatic cholestasis | [62] |
| Pakistani | NA * | NA * | NA * | c.822+1delG | NM_019050.2 | Homozygous | Splice site | Early-onset intrahepatic cholestasis | [62] |
| Pakistani | NA * | NA * | NA * | c.1214dupA p.Asn405fs | NM_019050.2 | Homozygous | Frameshift | Early-onset intrahepatic cholestasis | [62] |
| American | NA * | F | NA * | Duplication of 375 kb incl. entire gene + MYOZ2 + FABP2 (described at genomic DNA level) | ? | ? | Gross Ins/Dup | Cantu syndrome with coarse facial features | [63] |
| French–Canadian | NA * | NA * | NA * | 75 kb partial gene + FABP2 + C4orf3 (described at genomic DNA level) | ? | ? | Gross del | Confocal epilepsy | [64] |
4.5. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| α- | anti- |
| AMPAR | α-amino-3-hydroxy-5-methylisoxazolepropionate receptor |
| PBS | phosphate-buffered saline |
| BSA | bovine serum albumin |
| CNS | central nervous system |
| CNV | copy number variant |
| DSM | Diagnostic and Statistical Manual |
| EGTA | ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid |
| EDTA | ethylenediaminetetraacetic acid |
| GRIA | glutamate ionotropic receptor AMPA type |
| GRIP | glutamate receptor interacting protein |
| GluAR | glutamate AMPA receptor |
| GWAS | genome wide association study |
| LoF | loss of function |
| mGluR | metabotropic glutamate receptor |
| MAF | mean allele frequency |
| NMDA | N-methyl D-aspartate |
| PBST | phosphate-buffered saline with Triton X-100 |
| PFA | paraformaldehyde |
| PPI | protein-to-protein interaction |
| PVDE | polyvinylidene difluoride diethylene |
| SNV | single nucleotide variant |
| VOI | variant of interest |
| TBST | tris-buffered saline with Tween 80 |
| WES | whole exome sequencing |
| WGS | whole genome sequencing |
References
- Van Os, J.; Hanssen, M.; Bijl, R.V.; Vollebergh, W. Prevalence of Psychotic Disorder and Community Level of Psychotic Symptoms: An Urban-Rural Comparison. Arch. Gen. Psychiatry 2001, 58, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Goddard, M.E.; Derks, E.M.; Wray, N.R. Evidence-based psychiatric genetics, AKA the false dichotomy between common and rare variant hypotheses. Mol. Psychiatry 2012, 17, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Pardo, J.V. No Mendelian genes in psychiatry? J. Psychiatry Brain Sci. 2021, 6, e210019. [Google Scholar] [CrossRef]
- Lencz, T.; Yu, J.; Khan, R.R.; Flaherty, E.; Carmi, S.; Lam, M.; Ben-Avraham, D.; Barzilai, N.; Bressman, S.; Darvasi, A.; et al. Novel ultra-rare exonic variants identified in a founder population implicate cadherins in schizophrenia. Neuron 2021, 109, 1465–1478.e4. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, A.W.; Dhindsa, R.S.; Goldberg, T.E.; Mehralizade, A.; Motelow, J.E.; Wang, X.; Alkelai, A.; Harms, M.B.; Lieberman, J.A.; Markx, S.; et al. High-impact rare genetic variants in severe schizophrenia. Proc. Natl. Acad. Sci. USA 2021, 118, e2112560118. [Google Scholar] [CrossRef]
- Singh, T.; Poterba, T.; Curtis, D.; Akil, H.; Al Eissa, M.; Barchas, J.D.; Bass, N.; Bigdeli, T.B.; Breen, G.; Bromet, E.J.; et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature 2022, 604, 509–516. [Google Scholar] [CrossRef]
- Palmer, D.S.; Howrigan, D.P.; Chapman, S.B.; Adolfsson, R.; Bass, N.; Blackwood, D.; Boks, M.P.M.; Chen, C.Y.; Churchhouse, C.; Corvin, A.P.; et al. Exome sequencing in bipolar disorder identifies AKAP11 as a risk gene shared with schizophrenia. Nat. Genet. 2022, 54, 541–547. [Google Scholar] [CrossRef]
- Liu, D.; Meyer, D.A.; Fennessy, B.; Feng, C.; Cheng, E.; Johnson, J.S.; Park, Y.J.; Rieder, M.; Ascolillo, S.; de Pins, A.; et al. Schizophrenia risk conferred by rare protein-truncating variants is conserved across diverse human populations. Nat. Genet. 2023, 55, 369–376. [Google Scholar] [CrossRef]
- Nakamura, T.; Takata, A. The molecular pathology of schizophrenia: An overview of existing knowledge and new directions for future research. Mol. Psychiatry 2023. [Google Scholar] [CrossRef]
- Wolf, U. Identical mutations and phenotypic variation. Hum. Genet. 1997, 100, 305–321. [Google Scholar] [CrossRef]
- MacArthur, D.G.; Manolio, T.A.; Dimmock, D.P.; Rehm, H.L.; Shendure, J.; Abecasis, G.R.; Adams, D.R.; Altman, R.B.; Antonarakis, S.E.; Ashley, E.A.; et al. Guidelines for investigating causality of sequence variants in human disease. Nature 2014, 508, 469. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.; Berg, K.; Blehar, M.; Bowman, E.; Cloninger, R.C.; Depaulo, R.J.; Faraone, S.V.; Friedman, J.H.; Gershon, E.; Guroff, J.J.; et al. DIGS v4.0/SZ (Online Government Document); Bethesda, MD. Available online: https://www.nimhgenetics.org/data/resources/clinical-instruments/digs/v3.0/DIGS-v3.0-v4.0-version-notes.pdf (accessed on 5 October 2023).
- Helena, M.; Semple, R.J. DI-PAD CGP version 1.5 (Online Resource); Department of Psychiatry & the Behavioral Sciences, Keck School of Medicine, University of Southern California Los Angeles, CA 06/30/2011s. Available online: https://www.nimhgenetics.org/data/resources/clinical-instruments/other-instruments/DI-PAD/DI-PAD-CGP-v1.5.pdf (accessed on 5 October 2023).
- Kay, S.R.; Opler, L.A.; Spitzer, R.L.; Williams, J.B.; Fiszbein, A.; Gorelick, A. SCID-PANSS: Two-tier diagnostic system for psychotic disorders. Compr. Psychiatry 1991, 32, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M. Development of a rating scale for primary depressive illness. Br. J. Soc. Clin. Psychol. 1967, 6, 278–296. [Google Scholar] [CrossRef]
- Hamilton, M. The assessment of anxiety states by rating. Br. J. Med. Psychol. 1959, 32, 50–55. [Google Scholar] [CrossRef]
- Kanwal, A.; Sheikh, S.A.; Naz, S.; Pardo, J.V. Preliminary studies on apparent Mendelian psychotic disorders in consanguineous families. BMC Psychiatry 2022, 22, 709. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, A.; Pardo, J.V.; Naz, S. RGS3 and IL1RAPL1 missense variants implicate defective neurotransmission in early-onset inherited schizophrenias. J. Psychiatry Neurosci. 2022, 47, E379. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra, 1st ed.; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2020. [Google Scholar]
- Itan, Y.; Shang, L.; Boisson, B.; Patin, E.; Bolze, A.; Moncada-Velez, M.; Scott, E.; Ciancanelli, M.J.; Lafaille, F.G.; Markle, J.G.; et al. The human gene damage index as a gene-level approach to prioritizing exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 13615–13620. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Skolnick, J.; Gao, M.; Zhou, H.; Singh, S. AlphaFold 2: Why It Works and Its Implications for Understanding the Relationships of Protein Sequence, Structure, and Function. J. Chem. Inf. Model 2021, 61, 4827–4831. [Google Scholar] [CrossRef]
- Sheehan, D.V.; Lecrubier, Y.; Sheehan, K.H.; Amorim, P.; Janavs, J.; Weiller, E.; Hergueta, T.; Baker, R.; Dunbar, G.C. The Mini-International Neuropsychiatric Interview (M.I.N.I.): The development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J. Clin. Psychiatry 1998, 59 (Suppl. S20), 22–33, quiz 34–57. [Google Scholar] [PubMed]
- Harrison, P.J. The hippocampus in schizophrenia: A review of the neuropathological evidence and its pathophysiological implications. Psychopharmacology 2004, 174, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Schobel, S.A.; Chaudhury, N.H.; Khan, U.A.; Paniagua, B.; Styner, M.A.; Asllani, I.; Inbar, B.P.; Corcoran, C.M.; Lieberman, J.A.; Moore, H.; et al. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron 2013, 78, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, J.A.; Girgis, R.R.; Brucato, G.; Moore, H.; Provenzano, F.; Kegeles, L.; Javitt, D.; Kantrowitz, J.; Wall, M.M.; Corcoran, C.M.; et al. Hippocampal dysfunction in the pathophysiology of schizophrenia: A selective review and hypothesis for early detection and intervention. Mol. Psychiatry 2018, 23, 1764–1772. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, S.; Matsumoto, M.; van Erp, T.G.M. Hippocampal subregion abnormalities in schizophrenia: A systematic review of structural and physiological imaging studies. Neuropsychopharmacol. Rep. 2018, 38, 156–166. [Google Scholar] [CrossRef]
- Dong, H.; Zhang, P.; Song, I.; Petralia, R.S.; Liao, D.; Huganir, R.L. Characterization of the glutamate receptor-interacting proteins GRIP1 and GRIP2. J. Neurosci. 1999, 19, 6930–6941. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Krueger, R.F.; DeYoung, C.G. The RDoC initiative and the structure of psychopathology. Psychophysiology 2016, 53, 351–354. [Google Scholar] [CrossRef]
- Timms, A.E.; Dorschner, M.O.; Wechsler, J.; Choi, K.Y.; Kirkwood, R.; Girirajan, S.; Baker, C.; Eichler, E.E.; Korvatska, O.; Roche, K.W.; et al. Support for the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia from exome sequencing in multiplex families. JAMA Psychiatry 2013, 70, 582–590. [Google Scholar] [CrossRef]
- Knight, H.M.; Maclean, A.; Irfan, M.; Naeem, F.; Cass, S.; Pickard, B.S.; Muir, W.J.; Blackwood, D.H.; Ayub, M. Homozygosity mapping in a family presenting with schizophrenia, epilepsy and hearing impairment. Eur. J. Hum. Genet. 2008, 16, 750–758. [Google Scholar] [CrossRef]
- Xu, B.; Roos, J.L.; Dexheimer, P.; Boone, B.; Plummer, B.; Levy, S.; Gogos, J.A.; Karayiorgou, M. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat. Genet. 2011, 43, 864–868. [Google Scholar] [CrossRef]
- Fromer, M.; Pocklington, A.J.; Kavanagh, D.H.; Williams, H.J.; Dwyer, S.; Gormley, P.; Georgieva, L.; Rees, E.; Palta, P.; Ruderfer, D.M.; et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 2014, 506, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Blackstone, C.D.; Levey, A.I.; Huganir, R.L.; Price, D.L. Cellular localizations of AMPA glutamate receptors within the basal forebrain magnocellular complex of rat and monkey. J. Neurosci. 1993, 13, 2249–2263. [Google Scholar] [CrossRef] [PubMed]
- Quesada, V.; Diaz-Perales, A.; Gutierrez-Fernandez, A.; Garabaya, C.; Cal, S.; Lopez-Otin, C. Cloning and enzymatic analysis of 22 novel human ubiquitin-specific proteases. Biochem. Biophys. Res. Commun. 2004, 314, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Kaźmierczak, M.; Harris, S.L.; Kazmierczak, P.; Shah, P.; Starovoytov, V.; Ohlemiller, K.K.; Schwander, M. Progressive hearing loss in mice carrying a mutation in Usp53. J. Neurosci. 2015, 35, 15582–15598. [Google Scholar] [CrossRef]
- Alhebbi, H.; Peer-Zada, A.A.; Al-Hussaini, A.A.; Algubaisi, S.; Albassami, A.; AlMasri, N.; Alrusayni, Y.; Alruzug, I.M.; Alharby, E.; Samman, M.A.; et al. New paradigms of USP53 disease: Normal GGT cholestasis, BRIC, cholangiopathy, and responsiveness to rifampicin. J. Hum. Genet. 2021, 66, 151–159. [Google Scholar] [CrossRef]
- Subaran, R.L.; Odgerel, Z.; Swaminathan, R.; Glatt, C.E.; Weissman, M.M. Novel variants in ZNF34 and other brain-expressed transcription factors are shared among early-onset MDD relatives. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2016, 171B, 333–341. [Google Scholar] [CrossRef]
- Goes, F.S.; Pirooznia, M.; Tehan, M.; Zandi, P.P.; McGrath, J.; Wolyniec, P.; Nestadt, G.; Pulver, A.E. De novo variation in bipolar disorder. Mol. Psychiatry 2021, 26, 4127–4136. [Google Scholar] [CrossRef]
- Zhu, J.; Lee, K.Y.; Jewett, K.A.; Man, H.Y.; Chung, H.J.; Tsai, N.P. Epilepsy-associated gene Nedd4-2 mediates neuronal activity and seizure susceptibility through AMPA receptors. PLoS Genet. 2017, 13, e1006634. [Google Scholar] [CrossRef]
- Fu, A.K.; Hung, K.W.; Fu, W.Y.; Shen, C.; Chen, Y.; Xia, J.; Lai, K.O.; Ip, N.Y. APC(Cdh1) mediates EphA4-dependent downregulation of AMPA receptors in homeostatic plasticity. Nat. Neurosci. 2011, 14, 181–189. [Google Scholar] [CrossRef]
- Lussier, M.P.; Herring, B.E.; Nasu-Nishimura, Y.; Neutzner, A.; Karbowski, M.; Youle, R.J.; Nicoll, R.A.; Roche, K.W. Ubiquitin ligase RNF167 regulates AMPA receptor-mediated synaptic transmission. Proc. Natl. Acad. Sci. USA 2012, 109, 19426–19431. [Google Scholar] [CrossRef]
- Schwarz, L.A.; Hall, B.J.; Patrick, G.N. Activity-dependent ubiquitination of GluA1 mediates a distinct AMPA receptor endocytosis and sorting pathway. J. Neurosci. 2010, 30, 16718–16729. [Google Scholar] [CrossRef] [PubMed]
- Lussier, M.P.; Nasu-Nishimura, Y.; Roche, K.W. Activity-dependent ubiquitination of the AMPA receptor subunit GluA2. J Neurosci. 2011, 31, 3077–3081. [Google Scholar] [CrossRef] [PubMed]
- Pollak, T.A.; Rogers, J.P.; Nagele, R.G.; Peakman, M.; Stone, J.M.; David, A.S.; McGuire, P. Antibodies in the diagnosis, prognosis, and prediction of psychotic disorders. Schizophr. Bull. 2019, 45, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Farber, N.B. The NMDA receptor hypofunction model of psychosis. Ann. N. Y. Acad. Sci. 2003, 1003, 119–130. [Google Scholar] [CrossRef]
- Nicoletti, F.; Orlando, R.; Di Menna, L.; Cannella, M.; Notartomaso, S.; Mascio, G.; Iacovelli, L.; Matrisciano, F.; Fazio, F.; Caraci, F.; et al. Targeting mGlu Receptors for Optimization of Antipsychotic Activity and Disease-Modifying Effect in Schizophrenia. Front. Psychiatry 2019, 10, 49. [Google Scholar] [CrossRef]
- Mueller, T.M.; Meador-Woodruff, J.H. Post-translational protein modifications in schizophrenia. NPJ Schizophr. 2020, 6, 5. [Google Scholar] [CrossRef]
- Li, C.; Gulledge, A.T. NMDA receptors enhance the fidelity of synaptic integration. eNeuro 2021, 8. [Google Scholar] [CrossRef]
- Ojha, P.; Pal, S.; Bhattacharyya, S. Regulation of Metabotropic Glutamate Receptor Internalization and Synaptic AMPA Receptor Endocytosis by the Postsynaptic Protein Norbin. J. Neurosci. 2022, 42, 731–748. [Google Scholar] [CrossRef]
- Luza, S.; Opazo, C.M.; Bousman, C.A.; Pantelis, C.; Bush, A.I.; Everall, I.P. The ubiquitin proteasome system and schizophrenia. Lancet Psychiatry 2020, 7, 528–537. [Google Scholar] [CrossRef]
- Widagdo, J.; Guntupalli, S.; Jang, S.E.; Anggono, V. Regulation of AMPA receptor trafficking by protein ubiquitination. Front. Mol. Neurosci. 2017, 10, 347. [Google Scholar] [CrossRef]
- Gezdirici, A.; Kalaycik Sengul, O.; Dogan, M.; Ozguven, B.Y.; Akbulut, E. Biallelic Novel USP53 Splicing Variant Disrupting the Gene Function that Causes Cholestasis Phenotype and Review of the Literature. Mol. Syndromol. 2023, 13, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Porta, G.; Rigo, P.S.; Porta, A.; Pugliese, R.P.; Danesi, V.L.; Oliveira, E.; Borges, C.C.; Ribeiro, C.; Miura, I.K. Progressive familial intrahepatic cholestasis associated with USP53 gene mutation in a Brazilian child. J. Pediatr. Gastroenterol. Nutr. 2021, 72, 674–676. [Google Scholar] [CrossRef] [PubMed]
- Maddirevula, S.; Alhebbi, H.; Alqahtani, A.; Algoufi, T.; Alsaif, H.S.; Ibrahim, N.; Abdulwahab, F.; Barr, M.; Alzaidan, H.; Almehaideb, A. Identification of novel loci for pediatric cholestatic liver disease defined by KIF12, PPM1F, USP53, LSR, and WDR83OS pathogenic variants. Genet. Med. 2019, 21, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, Y.; Gong, J.Y.; Li, L.T.; Li, J.Q.; Zhang, M.H.; Lu, Y.; Xie, X.B.; Hong, Y.R.; Yu, Z.; et al. Low-GGT intrahepatic cholestasis associated with biallelic USP53 variants: Clinical, histological and ultrastructural characterization. Liver Int. 2020, 40, 1142–1150. [Google Scholar] [CrossRef]
- Bull, L.N.; Ellmers, R.; Foskett, P.; Strautnieks, S.; Sambrotta, M.; Czubkowski, P.; Jankowska, I.; Wagner, B.; Deheragoda, M.; Thompson, R.J. Cholestasis due to USP53 deficiency. J. Pediatr. Gastroenterol. Nutr. 2021, 72, 667. [Google Scholar] [CrossRef]
- Shatokhina, O.; Semenova, N.; Demina, N.; Dadali, E.; Polyakov, A.; Ryzhkova, O. A Two-Year Clinical Description of a Patient with a Rare Type of Low-GGT Cholestasis Caused by a Novel Variant of USP53. Genes 2021, 12, 1618. [Google Scholar] [CrossRef]
- Vij, M.; Sankaranarayanan, S. Biallelic Mutations in Ubiquitin-Specific Peptidase 53 (USP53) Causing Progressive Intrahepatic Cholestasis. Report of a Case with Review of Literature. Pediatr. Dev. Pathol. 2022, 25, 207–212. [Google Scholar] [CrossRef]
- Cheema, H.; Bertoli-Avella, A.M.; Skrahina, V.; Anjum, M.N.; Waheed, N.; Saeed, A.; Beetz, C.; Perez-Lopez, J.; Rocha, M.E.; Alawbathani, S. Genomic testing in 1019 individuals from 349 Pakistani families results in high diagnostic yield and clinical utility. NPJ Genom. Med. 2020, 5, 44. [Google Scholar] [CrossRef]
- Kurban, M.; Kim, C.; Kiuru, M.; Fantauzzo, K.; Cabral, R.; Abbas, O.; Levy, B.; Christiano, A.M. Copy number variations on chromosome 4q26–27 are associated with Cantu syndrome. Dermatology 2012, 223, 316–320. [Google Scholar] [CrossRef]
- Monlong, J.; Girard, S.L.; Meloche, C.; Cadieux-Dion, M.; Andrade, D.M.; Lafreniere, R.G.; Gravel, M.; Spiegelman, D.; Dionne-Laporte, A.; Boelman, C. Global characterization of copy number variants in epilepsy patients from whole genome sequencing. PLoS Genet. 2018, 14, e1007285. [Google Scholar] [CrossRef]
- Abu Rayyan, A.; Kamal, L.; Casadei, S.; Brownstein, Z.; Zahdeh, F.; Shahin, H.; Canavati, C.; Dweik, D.; Jaraysa, T.; Rabie, G.; et al. Genomic analysis of inherited hearing loss in the Palestinian population. Proc. Natl. Acad. Sci. USA 2020, 117, 20070–20076. [Google Scholar] [CrossRef] [PubMed]
- Alazami, A.M.; Patel, N.; Shamseldin, H.E.; Anazi, S.; Al-Dosari, M.S.; Alzahrani, F.; Hijazi, H.; Alshammari, M.; Aldahmesh, M.A.; Salih, M.A.; et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015, 10, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Grimberg, J.; Nawoschik, S.; Belluscio, L.; McKee, R.; Turck, A.; Eisenberg, A. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res. 1989, 17, 8390. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.Y.; Clark, J.M. Addition of a competitive primer can dramatically improve the specificity of PCR amplification of specific alleles. Biotechniques 1996, 21, 586. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanwal, A.; Sheikh, S.A.; Aslam, F.; Yaseen, S.; Beetham, Z.; Pankratz, N.; Clabots, C.R.; Naz, S.; Pardo, J.V. Genome Sequencing of Consanguineous Family Implicates Ubiquitin-Specific Protease 53 (USP53) Variant in Psychosis/Schizophrenia: Wild-Type Expression in Murine Hippocampal CA 1–3 and Granular Dentate with AMPA Synapse Interactions. Genes 2023, 14, 1921. https://doi.org/10.3390/genes14101921
Kanwal A, Sheikh SA, Aslam F, Yaseen S, Beetham Z, Pankratz N, Clabots CR, Naz S, Pardo JV. Genome Sequencing of Consanguineous Family Implicates Ubiquitin-Specific Protease 53 (USP53) Variant in Psychosis/Schizophrenia: Wild-Type Expression in Murine Hippocampal CA 1–3 and Granular Dentate with AMPA Synapse Interactions. Genes. 2023; 14(10):1921. https://doi.org/10.3390/genes14101921
Chicago/Turabian StyleKanwal, Ambreen, Sohail A. Sheikh, Faiza Aslam, Samina Yaseen, Zachary Beetham, Nathan Pankratz, Connie R. Clabots, Sadaf Naz, and José V. Pardo. 2023. "Genome Sequencing of Consanguineous Family Implicates Ubiquitin-Specific Protease 53 (USP53) Variant in Psychosis/Schizophrenia: Wild-Type Expression in Murine Hippocampal CA 1–3 and Granular Dentate with AMPA Synapse Interactions" Genes 14, no. 10: 1921. https://doi.org/10.3390/genes14101921