
Brain Gene Co-Expression Network Analysis Identifies 22q13 Region Genes Associated with Autism, Intellectual Disability, Seizures, Language Impairment, and Hypotonia


Abstract
:1. Introduction
2. Materials and Methods
2.1. Strategy
2.2. Genes Located on 22q13.2–q13.33
2.3. Genes Associated with Neurological Phenotypes
2.4. BrainSpan Gene Expression Data
2.5. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.6. Functional Term Enrichment Analysis of Co-Expression Modules
2.7. Prioritization of the Candidate Genes
3. Results
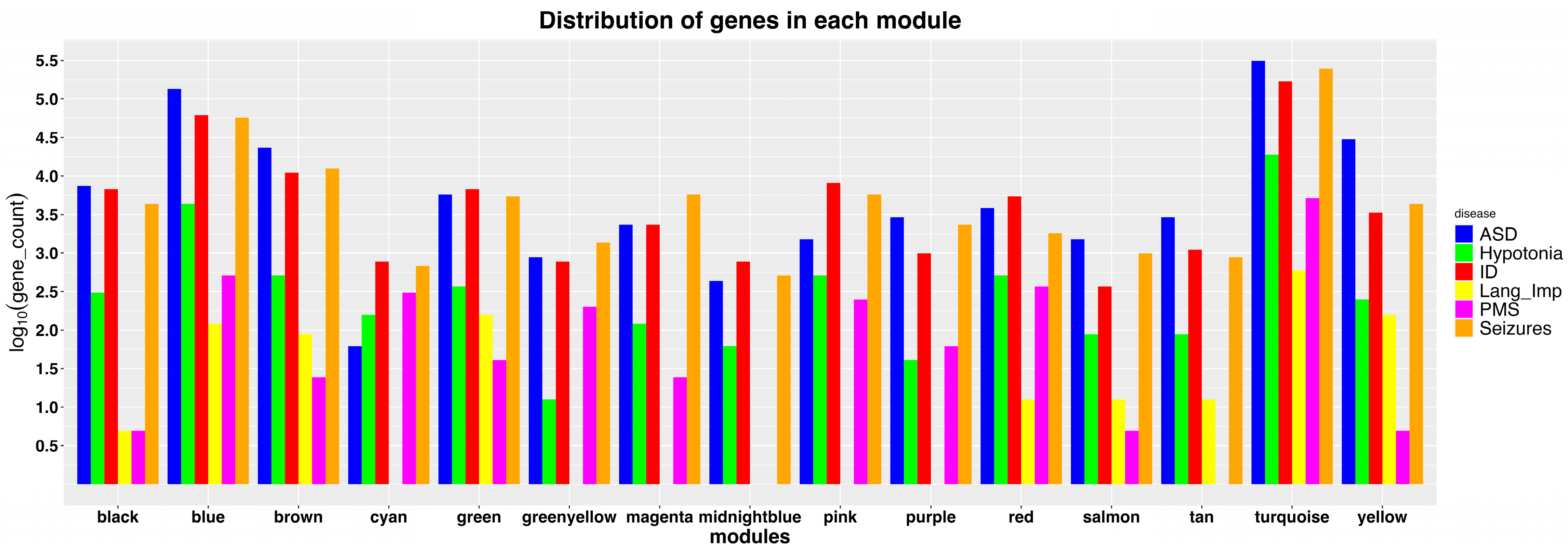
3.1. Identification of Modules Enriched with 22q13 Genes
3.2. ASD-Associated Candidate Genes in the 22q13.2–q13.33 Region
3.3. ID-Associated Candidate Genes in the 22q13.2–q13.33 Region
3.4. Seizure-Associated Candidate Genes in the 22q13.2–q13.33 Region
3.5. Candidate Genes Associated with Hypotonia and Language Impairment
3.6. Network Analysis to Reveal Candidate Gene Interactions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Phelan, K.; Mcdermid, H.E. The 22q13.3 Deletion Syndrome (Phelan-McDermid Syndrome). Mol. Syndr. 2012, 2, 186–201. [Google Scholar] [CrossRef]
- Soorya, L.; Kolevzon, A.; Zweifach, J.; Lim, T.; Dobry, Y.; Schwartz, L.; Frank, Y.; Wang, A.T.; Cai, G.; Parkhomenko, E.; et al. Prospective Investigation of Autism and Genotype-Phenotype Correlations in 22q13 Deletion Syndrome and SHANK3 Deficiency. Mol. Autism 2013, 4, 18:1–18:17. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; Siper, P.M.; Durkin, A.; Weissman, J.; Muratet, F.; Halpern, D.; Trelles, M.D.P.; Frank, Y.; Lozano, R.; Wang, A.T.; et al. Delineation of the Genetic and Clinical Spectrum of Phelan-McDermid Syndrome Caused by SHANK3 Point Mutations. Mol. Autism 2018, 9, 31:1–31:20. [Google Scholar] [CrossRef] [PubMed]
- Bonaglia, M.C.; Giorda, R.; Beri, S.; De Agostini, C.; Novara, F.; Fichera, M.; Grillo, L.; Galesi, O.; Vetro, A.; Ciccone, R.; et al. Molecular Mechanisms Generating and Stabilizing Terminal 22q13 Deletions in 44 Subjects with Phelan/McDermid Syndrome. PLoS Genet. 2011, 7, e1002173:1–e1002173:12. [Google Scholar] [CrossRef] [PubMed]
- Phelan, K.; Rogers, R.C.; Boccuto, L. Phelan-McDermid Syndrome; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; GeneReviews®: Seattle, WA, USA, 2018. [Google Scholar]
- Phelan, K.; Boccuto, L.; Powell, C.M.; Boeckers, T.M.; van Ravenswaaij-Arts, C.; Rogers, R.C.; Sala, C.; Verpelli, C.; Thurm, A.; Bennett, W.E., Jr.; et al. Phelan-McDermid Syndrome: A Classification System after 30 years of Experience. Orphanet J. Rare Dis. 2022, 17, 27:1–27:4. [Google Scholar] [CrossRef] [PubMed]
- Ricciardello, A.; Tomaiuolo, P.; Persico, A.M. Genotype–phenotype Correlation in Phelan-McDermid Syndrome: A Comprehensive Review of Chromosome 22q13 Deleted Genes. Am. J. Med. Genet. A 2021, 185, 2211–2233. [Google Scholar] [CrossRef] [PubMed]
- Mitz, A.R.; Philyaw, T.J.; Boccuto, L.; Shcheglovitov, A.; Sarasua, S.M.; Kaufmann, W.E.; Thurm, A. Identification of 22q13 Genes most Likely to Contribute to Phelan McDermid Syndrome. Eur. J. Hum. Genet. 2018, 26, 293–302. [Google Scholar] [CrossRef]
- Ziats, C.A.; Grosvenor, L.P.; Sarasua, S.M.; Thurm, A.E.; Swedo, S.E.; Mahfouz, A.; Rennert, O.M.; Ziats, M.N. Functional Genomics Analysis of Phelan-McDermid Syndrome 22q13 Region during Human Neurodevelopment. PLoS ONE 2019, 14, e0213921:1–e0213921:13. [Google Scholar] [CrossRef] [PubMed]
- Sarasua, S.M.; Dwivedi, A.; Boccuto, L.; Rollins, J.D.; Chen, C.; Rogers, R.C.; Phelan, K.; Dupont, B.R.; Collins, J.S. Association between Deletion Size and Important Phenotypes Expands the Genomic Region of Interest in Phelan-McDermid Syndrome (22q13 Deletion Syndrome). J. Med. Genet. 2011, 48, 761–766. [Google Scholar] [CrossRef]
- Vitrac, A.; Leblond, C.S.; Rolland, T.; Cliquet, F.; Mathieu, A.; Maruani, A.; Delorme, R.; Schön, M.; Grabrucker, A.M.; van Ravenswaaij-Arts, C.; et al. Dissecting the 22q13 Region to Explore the Genetic and Phenotypic Diversity of Patients with Phelan-McDermid Syndrome. Eur. J. Med. Genet. 2023, 66, 104732:1–104732:13. [Google Scholar] [CrossRef] [PubMed]
- Tabet, A.; Rolland, T.; Ducloy, M.; Lévy, J.; Buratti, J.; Mathieu, A.; Haye, D.; Perrin, L.; Dupont, C.; Passemard, S.; et al. A Framework to Identify Contributing Genes in Patients with Phelan-McDermid Syndrome. NPJ Genom. Med. 2017, 2, 32:1–32:9. [Google Scholar] [CrossRef] [PubMed]
- Samogy-Costa, C.I.; Varella-Branco, E.; Monfardini, F.; Ferraz, H.; Fock, R.A.; Barbosa, R.H.A.; Pessoa, A.L.S.; Perez, A.B.A.; Lourenço, N.; Vibranovski, M.; et al. A Brazilian Cohort of Individuals with Phelan-McDermid Syndrome: Genotype-Phenotype Correlation and Identification of an Atypical Case. J. Neurodev. Disord. 2019, 11, 13:1–13:10. [Google Scholar] [CrossRef] [PubMed]
- Levy, T.; Foss-Feig, J.H.; Betancur, C.; Siper, P.M.; Trelles-Thorne, M.d.P.; Halpern, D.; Frank, Y.; Lozano, R.; Layton, C.; Britvan, B.; et al. Strong Evidence for Genotype–phenotype Correlations in Phelan-McDermid Syndrome: Results from the Developmental Synaptopathies Consortium. Hum. Mol. Genet. 2022, 31, 625–637. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559:1–559:13. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.A.; Ding, S.; Sunkin, S.M.; Smith, K.A.; Ng, L.; Szafer, A.; Ebbert, A.; Riley, Z.L.; Royall, J.J.; Aiona, K.; et al. Transcriptional Landscape of the Prenatal Human Brain. Nature 2014, 508, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3:1–P3:17. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Cogill, S.; Wang, L. Support Vector Machine Model of Developmental Brain Gene Expression Data for Prioritization of Autism Risk Gene Candidates. Bioinformatics 2016, 32, 3611–3618. [Google Scholar] [CrossRef]
- Gudenas, B.L.; Wang, L. Gene Coexpression Networks in Human Brain Developmental Transcriptomes Implicate the Association of Long Noncoding RNAs with Intellectual Disability. Bioinform. Biol. Insights 2015, 9, 21–27. [Google Scholar] [CrossRef]
- Willsey, A.J.; Sanders, S.J.; Li, M.; Dong, S.; Tebbenkamp, A.T.; Muhle, R.A.; Reilly, S.K.; Lin, L.; Fertuzinhos, S.; Miller, J.A.; et al. Coexpression Networks Implicate Human Midfetal Deep Cortical Projection Neurons in the Pathogenesis of Autism. Cell 2013, 155, 997–1007. [Google Scholar] [CrossRef]
- Guo, Y.; Ma, J.; Xiao, L.; Fang, J.; Li, G.; Zhang, L.; Xu, L.; Lai, X.; Pan, G.; Chen, Z. Identification of Key Pathways and Genes in Different Types of Chronic Kidney Disease Based on WGCNA. Mol. Med. Rep. 2019, 20, 2245–2257. [Google Scholar] [CrossRef]
- Niu, X.; Zhang, J.; Zhang, L.; Hou, Y.; Pu, S.; Chu, A.; Bai, M.; Zhang, Z. Weighted Gene Co-Expression Network Analysis Identifies Critical Genes in the Development of Heart Failure after Acute Myocardial Infarction. Front. Genet. 2019, 10, 1214:1–1214:17. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Su, R.; Liao, Y.; Li, Y.; Li, L. Identification of 10 Hub Genes Related to the Progression of Colorectal Cancer by Co-Expression Analysis. PeerJ 2020, 8, e9633:1–e9633:16. [Google Scholar] [CrossRef] [PubMed]
- UCSC Genome Browser Gateway. Available online: https://genome.ucsc.edu/goldenPath/releaseLog.html#latest (accessed on 18 September 2023).
- Banerjee-Basu, S.; Packer, A. SFARI Gene: An Evolving Database for the Autism Research Community. Dis. Model. Mech. 2010, 3, 133–135. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, D.; Xue, H.; Liu, X.; Zhang, T.; Tang, S.; Li, W.; Xu, X. IDGenetics: A Comprehensive Database for Genes and Mutations of Intellectual Disability Related Disorders. Neurosci. Lett. 2018, 685, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lin, Z.; Liu, L.; Xu, H.; Shi, Y.; Yi, Y.; He, N.; Liao, W. Epilepsy-Associated Genes. Seizure 2017, 44, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.R.; Hem, V.; Katz, K.S.; Ovetsky, M.; Wallin, C.; Ermolaeva, O.; Tolstoy, I.; Tatusova, T.; Pruitt, K.D.; Maglott, D.R.; et al. Gene: A Gene-Centered Information Resource at NCBI. Nucleic Acids Res. 2015, 43, D36–D42. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE Reference Annotation for the Human and Mouse Genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef]
- Li, J.; Zhou, D.; Qiu, W.; Shi, Y.; Yang, J.J.; Chen, S.; Wang, Q.; Pan, H. Application of Weighted Gene Co-Expression Network Analysis for Data from Paired Design. Sci. Rep. 2018, 8, 622:1–622:8. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. the Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Wilson, H.L.; Wong, A.C.C.; Shaw, S.R.; Tse, W.Y.; Stapleton, G.A.; Phelan, M.C.; Hu, S.; Marshall, J.; McDermid, H.E. Molecular Characterisation of the 22q13 Deletion Syndrome Supports the Role of Haploinsufficiency of SHANK3/PROSAP2 in the Major Neurological Symptoms. J. Med. Genet. 2003, 40, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Lv, H.; Yang, T.; Du, X.; Sun, Y.; Xiao, B.; Fan, Y.; Luo, X.; Zhan, Y.; Wang, L.; et al. A 29 Mainland Chinese Cohort of Patients with Phelan–McDermid Syndrome: Genotype–phenotype Correlations and the Role of SHANK3 Haploinsufficiency in the Important Phenotypes. Orphanet J. Rare Dis. 2020, 15, 335:1–335:12. [Google Scholar] [CrossRef]
- Lutz, A.K.; Pfaender, S.; Incearap, B.; Ioannidis, V.; Ottonelli, I.; Föhr, K.J.; Cammerer, J.; Zoller, M.; Higelin, J.; Giona, F.; et al. Autism-Associated SHANK3 Mutations Impair Maturation of Neuromuscular Junctions and Striated Muscles. Sci. Transl. Med. 2020, 12, eaaz3267:1–eaaz3267:15. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Kasper, L.H.; Bedford, D.C.; Lerach, S.; Teubner, B.J.W.; Brindle, P.K. Mutation of the CH1 Domain in the Histone Acetyltransferase CREBBP Results in Autism-Relevant Behaviors in Mice. PLoS ONE 2016, 11, e0146366:1–e0146366:18. [Google Scholar] [CrossRef]
- Simenson, K.; Õiglane-Shlik, E.; Teek, R.; Kuuse, K.; Õunap, K. A Patient with the Classic Features of Phelan-McDermid Syndrome and a High Immunoglobulin E Level Caused by a Cryptic Interstitial 0.72-Mb Deletion in the 22q13. 2 Region. Am. J. Med. Genet. A 2014, 164, 806–809. [Google Scholar] [CrossRef]
- Wilson, H.L.; Crolla, J.A.; Walker, D.; Artifoni, L.; Dallapiccola, B.; Takano, T.; Vasudevan, P.; Huang, S.; Maloney, V.; Yobb, T.; et al. Interstitial 22q13 Deletions: Genes Other than SHANK3 have Major Effects on Cognitive and Language Development. Eur. J. Hum. Genet. 2008, 16, 1301–1310. [Google Scholar] [CrossRef]
- Fujita, Y.; Mochizuki, D.; Mori, Y.; Nakamoto, N.; Kobayashi, M.; Omi, K.; Kodama, H.; Yanagawa, Y.; Abe, T.; Tsuzuku, T.; et al. Girl with Accelerated Growth, Hearing Loss, Inner Ear Anomalies, Delayed Myelination of the Brain, and Del(22)(q13.1q13.2). Am. J. Med. Genet. 2000, 92, 195–199. [Google Scholar] [CrossRef]
- Romain, D.R.; Goldsmith, J.; Cairney, H.; Columbano-Green, L.M.; Smythe, R.H.; Parfitt, R.G. Partial Monosomy for Chromosome 22 in a Patient with Del(22)(Pter----q13.1::q13.33----Qter). J. Med. Genet. 1990, 27, 588–589. [Google Scholar] [CrossRef]
- Disciglio, V.; Rizzo, C.L.; Mencarelli, M.A.; Mucciolo, M.; Marozza, A.; Di Marco, C.; Massarelli, A.; Canocchi, V.; Baldassarri, M.; Ndoni, E.; et al. Interstitial 22q13 Deletions Not Involving SHANK3 Gene: A New Contiguous Gene Syndrome. Am. J. Med. Genet. A 2014, 164, 1666–1676. [Google Scholar] [CrossRef]
- Sarasua, S.M.; Boccuto, L.; Sharp, J.L.; Dwivedi, A.; Chen, C.F.; Rollins, J.D.; Rogers, R.C.; Phelan, K.; DuPont, B.R. Clinical and Genomic Evaluation of 201 Patients with Phelan–McDermid Syndrome. Hum. Genet. 2014, 133, 847–859. [Google Scholar] [CrossRef]
- Nevado, J.; García-Miñaúr, S.; Palomares-Bralo, M.; Vallespín, E.; Guillén-Navarro, E.; Rosell, J.; Bel-Fenellós, C.; Mori, M.Á.; Milá, M.; Campo, M.D.; et al. Variability in Phelan-McDermid Syndrome in a Cohort of 210 Individuals. Front. Genet. 2022, 13, 652454:1–652454:24. [Google Scholar] [CrossRef]
- Stevens, C.A. Rubinstein-Taybi Syndrome; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; GeneReviews®: Seattle, WA, USA, 2019. [Google Scholar]
- Fergelot, P.; Van Belzen, M.; Van Gils, J.; Afenjar, A.; Armour, C.M.; Arveiler, B.; Beets, L.; Burglen, L.; Busa, T.; Collet, M.; et al. Phenotype and Genotype in 52 Patients with Rubinstein–Taybi Syndrome Caused by EP300 Mutations. Am. J. Med. Genet. A 2016, 170, 3069–3082. [Google Scholar] [CrossRef]
- Gayther, S.A.; Batley, S.J.; Linger, L.; Bannister, A.; Thorpe, K.; Chin, S.F.; Daigo, Y.; Russell, P.; Wilson, A.; Sowter, H.M.; et al. Mutations Truncating the EP300 Acetylase in Human Cancers. Nat. Genet. 2000, 24, 300–303. [Google Scholar] [CrossRef]
- Van Gils, J.; Magdinier, F.; Fergelot, P.; Lacombe, D. Rubinstein-Taybi Syndrome: A Model of Epigenetic Disorder. Genes 2021, 12, 968. [Google Scholar] [CrossRef] [PubMed]
- Krauss, G. Signal Transmission via Transmembrane Receptors with Tyrosine-Specific Protein Kinase Activity. In Biochemistry of Signal Transduction and Regulation, 5th ed.; Krauss, G., Ed.; Wiley-VCH: Bayreuth, Germany, 2004; pp. 473–533. ISBN 978-352-766-747-5. [Google Scholar]
- Potocki, L.; Bi, W.; Treadwell-Deering, D.; Carvalho, C.M.B.; Eifert, A.; Friedman, E.M.; Glaze, D.; Krull, K.; Lee, J.A.; Lewis, R.A.; et al. Characterization of Potocki-Lupski Syndrome (Dup(17)(p11.2p11.2)) and Delineation of a Dosage-Sensitive Critical Interval that can Convey an Autism Phenotype. Am. J. Hum. Genet. 2007, 80, 633–649. [Google Scholar] [CrossRef] [PubMed]
- Walz, K.; Paylor, R.; Yan, J.; Bi, W.; Lupski, J.R. Rai1 Duplication Causes Physical and Behavioral Phenotypes in a Mouse Model of Dup(17)(p11.2p11.2). J. Clin. Investig. 2006, 116, 3035–3041. [Google Scholar] [CrossRef] [PubMed]
- Jerath, N.U.; Shy, M.E. Hereditary Motor and Sensory Neuropathies: Understanding Molecular Pathogenesis could Lead to Future Treatment Strategies. Biochim. Biophys. Acta 2015, 1852, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Abid, I.; Kamoun, F.; Boubaker, C.; Delague, V.; Triki, C. Epilepsy in Patients with Charcot Marie Tooth Disease: Phenotypic Spectrum Or Two Different Diseases? J. Pediatr. Epilepsy 2014, 3, 121–125. [Google Scholar] [CrossRef]
- Monteiro, I.; Jesus-Ribeiro, J.; Melo, J.B.; Sales, F.; Geraldo, A. Generalized Epilepsy in a Patient with GBJ1 X-Linked Charcot-Marie-Tooth Disease. Sinapse 2022, 22, 3:1–3:5. [Google Scholar] [CrossRef]
- Tsao, C.Y. Intractable Epilepsy, Audio-Visual Hallucinations and Charcot-Marie-Tooth Disease 1A in an African-American Boy. Clin. EEG Neurosci. 2011, 42, 206–208. [Google Scholar] [CrossRef]
- Chaudhuri, J.; Dutta, A.K.; Biswas, T.; Biswas, A.; Ray, B.K.; Ganguly, G. Charcot-Marie-Tooth Disease Type 4J with Spastic Quadriplegia, Epilepsy and Global Developmental Delay: A Tale of Three Siblings. Int. J. Neurosci. 2022, 132, 783–786. [Google Scholar] [CrossRef]
- Sánchez-Jiménez, P.; Elizalde-Horcada, M.; Sanz-García, A.; Granero-Cremades, I.; De Toledo, M.; Pulido, P.; Navas, M.; Gago-Veiga, A.B.; Alonso-Guirado, L.; Alonso-Cerezo, M.C.; et al. DNA Methylation Description of Hippocampus, Cortex, Amygdala, and Blood of Drug-Resistant Temporal Lobe Epilepsy. Mol. Neurobiol. 2023, 60, 2070–2085. [Google Scholar] [CrossRef]
- Fryland, T.; Christensen, J.H.; Pallesen, J.; Mattheisen, M.; Palmfeldt, J.; Bak, M.; Grove, J.; Demontis, D.; Blechingberg, J.; Ooi, H.S.; et al. Identification of the BRD1 Interaction Network and its Impact on Mental Disorder Risk. Genome Med. 2016, 8, 53:1–53:20. [Google Scholar] [CrossRef]
- Severinsen, J.E.; Bjarkam, C.R.; Kiaer-Larsen, S.; Olsen, I.M.; Nielsen, M.M.; Blechingberg, J.; Nielsen, A.L.; Holm, I.E.; Foldager, L.; Young, B.D.; et al. Evidence Implicating BRD1 with Brain Development and Susceptibility to both Schizophrenia and Bipolar Affective Disorder. Mol. Psychiatry 2006, 11, 1126–1138. [Google Scholar] [CrossRef]
- Paternoster, V.; Edhager, A.V.; Qvist, P.; Donskov, J.G.; Shliaha, P.; Jensen, O.N.; Mors, O.; Nielsen, A.L.; Børglum, A.D.; Palmfeldt, J.; et al. Inactivation of the Schizophrenia-Associated BRD1 Gene in Brain Causes Failure-to-Thrive, Seizure Susceptibility and Abnormal Histone H3 Acetylation and N-Tail Clipping. Mol. Neurobiol. 2021, 58, 4495–4505. [Google Scholar] [CrossRef] [PubMed]
- Qvist, P.; Rajkumar, A.P.; Redrobe, J.P.; Nyegaard, M.; Christensen, J.H.; Mors, O.; Wegener, G.; Didriksen, M.; Børglum, A.D. Mice Heterozygous for an Inactivated Allele of the Schizophrenia Associated Brd1 Gene Display Selective Cognitive Deficits with Translational Relevance to Schizophrenia. Neurobiol. Learn. Mem. 2017, 141, 44–52. [Google Scholar] [CrossRef]
- Qvist, P.; Christensen, J.H.; Vardya, I.; Rajkumar, A.P.; Mørk, A.; Paternoster, V.; Füchtbauer, E.M.; Pallesen, J.; Fryland, T.; Dyrvig, M.; et al. The Schizophrenia-Associated BRD1 Gene Regulates Behavior, Neurotransmission, and Expression of Schizophrenia Risk Enriched Gene Sets in Mice. Biol. Psychiatry 2017, 82, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Paternoster, V.; Svanborg, M.; Edhager, A.V.; Rajkumar, A.P.; Eickhardt, E.A.; Pallesen, J.; Grove, J.; Qvist, P.; Fryland, T.; Wegener, G.; et al. Brain Proteome Changes in Female Brd1+/− Mice Unmask Dendritic Spine Pathology and show Enrichment for Schizophrenia Risk. Neurobiol. Dis. 2019, 124, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, A.P.; Qvist, P.; Donskov, J.G.; Lazarus, R.; Pallesen, J.; Nava, N.; Winther, G.; Liebenberg, N.; Cour, S.H.l.; Paternoster, V.; et al. Reduced Brd1 Expression Leads to Reversible Depression-Like Behaviors and Gene-Expression Changes in Female Mice. Transl. Psychiatry 2020, 10, 239:1–239:14. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes in the 22q13 Region | Functional Annotation | Associated Neurological Phenotype | Degree of Connectivity (%) | pLI Score |
|---|---|---|---|---|
| EP300 | Transcriptional regulation, chromatin modification, DNA repair | ASD | 48.99 | 1 |
| TCF20 | Regulation of transcription by RNA polymerase II | ASD | 48.46 | 1 |
| RBX1 | DNA damage response, negative regulation of Wnt signaling pathway | ASD | 45.16 | 0.95 |
| C22orf32 (SMDT1) | Mitochondrial ion transport | ASD | 43.57 | 0.18 |
| XPNPEP3 | Protein Maturation, proteolysis | ASD | 43.57 | 0 |
| PMM1 | Metabolic process, Glycosylation | ID | 28.53 | 0 |
| LMF2 | - | ID | 16.23 | 0 |
| SCO2 | Sensory organ development, Mitochondrial cytochrome C oxidase assembly | ID | 15.31 | 0 |
| C22orf40 (CDPF1) | - | ID | 13.35 | 0 |
| CHKB | Metabolic processes | ID | 12.04 | 0 |
| SREBF2 | Signaling pathway, negative regulation of DNA-templated transcription | Seizures | 33.54 | 0.21 |
| BRD1 | Regulation of transcription by RNA polymerase II, Regulation of DNA-templated transcription | Seizures | 31.91 | 1 |
| RANGAP1 | Activation of GTPase activity, maintenance of protein in nucleus, nuclear export, nuclear transport | Seizures | 31.29 | 0.03 |
| PPP6R2 | Regulation of phosphatase activity | Seizures | 29.54 | 0.16 |
| SBF1 | Protein dephosphorylation | Seizures | 25.53 | 1 |
| SHANK3 | Brain development, brain morphogenesis, Cell junction assembly | ASD, Seizures | 17.9 | 1 |
| NUP50 | Nuclear transport, protein localization to nucleus | Language impairment | 39.19 | 0.28 |
| TUBGCP6 | Cytoplasmic microtubule organization, spindle assembly | Language impairment | 26.09 | 0 |
| ST13 | Post-translational protein folding | Language impairment | 14.49 | 0.66 |
| ARSA | Intracellular lumen | Language impairment | 13.04 | 0 |
| CPT1B | Fatty acid metabolic process | Language impairment | 13.04 | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, S.; Sarasua, S.M.; Boccuto, L.; Dean, B.C.; Wang, L. Brain Gene Co-Expression Network Analysis Identifies 22q13 Region Genes Associated with Autism, Intellectual Disability, Seizures, Language Impairment, and Hypotonia. Genes 2023, 14, 1998. https://doi.org/10.3390/genes14111998
Shah S, Sarasua SM, Boccuto L, Dean BC, Wang L. Brain Gene Co-Expression Network Analysis Identifies 22q13 Region Genes Associated with Autism, Intellectual Disability, Seizures, Language Impairment, and Hypotonia. Genes. 2023; 14(11):1998. https://doi.org/10.3390/genes14111998
Chicago/Turabian StyleShah, Snehal, Sara M. Sarasua, Luigi Boccuto, Brian C. Dean, and Liangjiang Wang. 2023. "Brain Gene Co-Expression Network Analysis Identifies 22q13 Region Genes Associated with Autism, Intellectual Disability, Seizures, Language Impairment, and Hypotonia" Genes 14, no. 11: 1998. https://doi.org/10.3390/genes14111998