
The Interplay between T Cells and Cancer: The Basis of Immunotherapy

1
Department of Molecular Microbiology and Immunology, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA
2
Department of Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA
3
Department of Pediatrics, Children’s Hospital Los Angeles, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Genes 2023, 14(5), 1008; https://doi.org/10.3390/genes14051008
Submission received: 1 March 2023
/
Revised: 17 April 2023
/
Accepted: 24 April 2023
/
Published: 28 April 2023
(This article belongs to the Special Issue Cell Signalling and Inflammation in Cancer)
Abstract
:Over the past decade, immunotherapy has emerged as one of the most promising approaches to cancer treatment. The use of immune checkpoint inhibitors has resulted in impressive and durable clinical responses in the treatment of various cancers. Additionally, immunotherapy utilizing chimeric antigen receptor (CAR)-engineered T cells has produced robust responses in blood cancers, and T cell receptor (TCR)-engineered T cells are showing promising results in the treatment of solid cancers. Despite these noteworthy advancements in cancer immunotherapy, numerous challenges remain. Some patient populations are unresponsive to immune checkpoint inhibitor therapy, and CAR T cell therapy has yet to show efficacy against solid cancers. In this review, we first discuss the significant role that T cells play in the body’s defense against cancer. We then delve into the mechanisms behind the current challenges facing immunotherapy, starting with T cell exhaustion due to immune checkpoint upregulation and changes in the transcriptional and epigenetic landscapes of dysfunctional T cells. We then discuss cancer-cell-intrinsic characteristics, including molecular alterations in cancer cells and the immunosuppressive nature of the tumor microenvironment (TME), which collectively facilitate tumor cell proliferation, survival, metastasis, and immune evasion. Finally, we examine recent advancements in cancer immunotherapy, with a specific emphasis on T-cell-based treatments.
Keywords:
cancer; T cells; signaling; alterations; cell therapy; vaccine; antibody; CRISPR; CAR T/TCR T/TIL1. Introduction
Cytotoxic T lymphocytes (CTLs), or cytotoxic T cells, play a critical role in the recognition and elimination of tumor cells [1]. Cytotoxic T cells are activated via a two-step process. First, T cell receptors (TCRs) bind to tumor-derived peptides presented on major histocompatibility complex (MHC) molecules by antigen-presenting cells (APCs) [1,2]. This initial interaction establishes the antigen specificity of the T cell response. The second (co-stimulatory) T cell activation signal is mediated by the CD28–B7 interaction [1,2]. The combination of these two signals facilitates the full activation of cytotoxic T cells, enabling them to target and eliminate cells displaying their cognate antigen, including cancer cells, through antigen-specific killing [1,2]. Various cell-based therapies utilize genetically modified cell-surface antigen-specific receptors that aim to enhance and optimize the targeting and cytotoxic activity of T cells [3,4]. For example, TCR T cells and chimeric antigen receptor (CAR) T cells are examples of genetically engineered T cell therapies that have generated durable responses in clinical trials and demonstrate promising antitumor activity [3]. As of 2022, the first United States Food and Drug Administration (FDA)-approved TCR T therapy was granted approval for the treatment of HLA-A*02:01-positive uveal melanoma [5]. Various other TCR T cell clinical trials are currently underway and have shown promising results [3]. CAR T cells have likewise demonstrated impressive antitumor activities. Six CAR T cell products have been approved by the FDA, four of which target CD19-positive tumors and two of which target B cell maturation antigen (BCMA) [4,6,7,8]. A summary of ongoing or completed clinical trials of CAR T and TCR T therapies can be found in Table 1. Although the use of CAR T cells has achieved remarkable success in the treatment of hematological malignancies, poor clinical responses have been observed against solid tumors [4]. Various barriers are currently faced by cancer immunotherapy, including the unique properties of tumor cells that promote growth and immune evasion, the dysfunction of T cells that prevent the effective elimination of tumor cells, and an intricate interplay between tumor cells and T cells in the TME [9]. These obstacles must be overcome to fully harness the potential of cancer immunotherapy [10,11,12,13].
1.1. T Cell Receptor Structure and T Cell Signaling
In the early 1980s, studies utilizing immunological and biochemical techniques uncovered the composition and structure of the TCR complex [36,37,38]. The TCR is a heterodimer composed of two chains: α- and β-chains or γ- and δ-chains [39]. In humans, αβ T cells comprise 95% of all T cells and are responsible for recognizing peptides bound to MHC molecules [39]. The remaining 5% of the T cell population is composed of γδ T cells, which have the unique ability to function in MHC-independent antigen recognition [39]. The complete αβ TCR complex requires four additional CD3 polypeptide chains—the epsilon (ε), gamma (γ), delta (δ), and zeta (ζ) chains—that are critical to carrying out signal transduction initiated by TCR ligation [40]. These chains assemble as three pairs of dimers (γε, δε, and ζζ) to form a stable octameric complex with the αβ TCR, creating a complex with equal stoichiometric proportions of TCRαβ, CD3γε, CD3δε, and CD3ζζ [40].
Upon binding to the peptide–MHC complex, the conformation of the TCR complex changes, triggering lymphocyte-specific protein tyrosine kinase (Lck)-mediated phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) within the CD3 adaptor proteins [41]. pITAMs then serve as docking sites for zeta-chain-associated protein kinase 70 (Zap70), which binds via its tandem Src homology 2 (SH2) domains, resulting in its phosphorylation and activation [42,43,44]. Once activated, Zap70 phosphorylates multiple tyrosine residues in the linker for the activation of T cells (LAT), a key scaffold protein that recruits various downstream adaptor molecules and signaling proteins, including the SH-2-domain-containing leukocyte protein of 76 kDa (SLP-76), phospholipase Cγ1 (PLCγ1), son of sevenless (SOS), growth factor receptor-bound protein 2 (GRB2), interleukin (IL)-2-induced tyrosine kinase (ITK), Vav, non-catalytic tyrosine kinase (NCK), adhesion- and degranulation-promoting adapter protein (ADAP), and Grb2-related adaptor downstream of Shc (GADS) [45,46]. These signaling molecules activate the downstream IP3-Ca2+-NFAT, DAG-PKCθ-NF-κB, and DAG-RAS-AP1 pathways, which work together to promote T cell activation, proliferation, survival, and effector function [42,43,44]. Homeostatic regulation of these cornerstone pathways is critical for normal T cell function. However, various factors within the TME contribute to the dysregulation of these crucial pathways, leading to the impairment of T cell effector functions and cancer immune evasion.
1.2. Aberrant T Cell Signaling Associated with Various Malignancies
Dysfunctional T cell signaling has been implicated in multiple malignancies, including head and neck squamous cell carcinoma (HNSCC) [47,48], lung cancer (including non-small-cell lung cancer (NSCLC)) [49,50,51,52], hepatocellular carcinoma (HCC) [47,53,54,55,56,57], gastric cancer [56,58], T cell acute lymphoblastic leukemia (T-ALL) [59], chronic lymphocytic leukemia (CLL) [60,61], melanoma [62,63,64], lymphoma [65,66,67,68,69,70,71,72,73,74,75,76,77,78,79], colon cancer, and others [80]. The causes of these malignancies and their resistance to immunotherapy are multifaceted and can be attributed to altered expression of inhibitory receptors and negative regulators, as well as changes in the transcriptional and epigenetic landscapes of T cells. These changes ultimately result in a dysfunctional phenotype characterized by a loss of T cell effector functions and reduced antitumor efficacy. For instance, reduced interferon-gamma (IFNγ) production is often observed in the tumor-infiltrating lymphocytes (TILs) of patients with different forms of cancer, including melanoma [62], HNSCC [47], and ovarian cancer [81]. Attenuated tumor necrosis factor (TNF) production is seen in T cells isolated from mice with colon carcinoma [80] and in TILs of HNSCC [47] and ovarian cancer [81] patients. High granzyme K levels are observed in dysfunctional CTL populations of melanoma [82], HCC [54], and NSCLC patients [52]. Reduced perforin and granzyme B can also be observed in certain tumor-infiltrating CTLs [62,80].
2. Mechanisms Responsible for the Current Challenges Faced in Immunotherapy
2.1. Exhaustion and Dysfunction of T Cells
The imperative role of immune protection against cancer highlights the importance of functional T cell signaling and activity in preventing and combating cancer. T cell exhaustion, a hypofunctional T cell state characterized by a loss of self-renewal capacity and reduced effector function, is a significant contributor to cancer progression and constitutes a major obstacle in the successful application of CAR T cell therapy against solid cancers [83,84,85]. The cause of T cell exhaustion in the context of cancer is multifaceted, with persistent antigenic stimulation, the immune-inhibitory nature of the TME, changes in the T-cell-related transcriptional and epigenetic landscapes, and metabolic factors all identified as contributing factors [86].
T cell exhaustion is commonly characterized by an increased expression of inhibitory receptors [84]. Under normal physiological conditions, these inhibitory checkpoints serve a protective function. Ligation of inhibitory receptors (expressed on activated T cells) with their respective ligands (expressed on tissues and APCs) attenuates T cell responses and promotes T cell tolerance, thereby modulating inflammation and preventing collateral damage during chronic infections [87]. However, many tumors take advantage of this protective mechanism by overexpressing inhibitory ligands, which in turn ligate with inhibitory receptors. This triggers intracellular pathways that effectively prevent T cell effector functions against the tumor and consequently results in immune tolerance of the cancer [87].
2.1.1. Elevated Immune Checkpoint Expression Correlates with Poor Patient Outcomes and Reduced Responses to Cancer Immunotherapy
Heightened expression and ligation of inhibitory receptors, such as programmed cell death protein 1 (PD-1), T cell immunoglobulin and ITIM domain (TIGIT), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), lymphocyte activation gene 3 (LAG-3), T cell immunoglobulin and mucin domain 3 (TIM-3), B and T lymphocyte attenuator (BTLA), CD244, and CD160, upon chronic antigen exposure in various solid cancers results in limited T cell survival and function [11,12,13,47,48,51,54,56,63,81,82,84,88,89,90,91,92,93,94,95,96,97,98,99]. Furthermore, when compared to peripheral blood lymphocytes, TILs of solid cancers demonstrate higher expression of inhibitory receptors and a more dysregulated phenotype [81,90]. Notably, expression of PD-1 on T cells correlates with higher co-expression of other inhibitory receptors and a more exhausted phenotype [47,63]. In HNSCC patients, high expression of TIM-3, an immune checkpoint receptor that activates cell proliferation via the Akt/S6 pathway, is a marker of exhausted TILs [48]. Interestingly, TIM-3 expression alone does not dictate TIL exhaustion [47]. While increased expression of TIM-3 is a marker of a more extensively exhausted phenotype, co-expression with PD-1 is required for greater TIL dysfunction, further emphasizing the essential role of PD-1 in T cell exhaustion [47]. High expression of PD-1 in both TIM-3– and TIM-3+ TILs is associated with higher levels of B-lymphocyte-induced maturation protein-1 (BLIMP-1 and basic leucine zipper ATF-like transcription factor (BATF), transcription factors that impair T cell proliferation and cytokine secretion [47]. Co-expression of these receptors results in cross-talk that dampens Akt/S6 phosphorylation and regulates antitumor T cell responses [47,48].
It is evident that increased expression of inhibitory checkpoints contributes to T cell exhaustion of endogenous tumor-reactive T cells and therefore impacts the intrinsic antitumor capability of T cells. In addition, it has been found that elevated expression of negative regulators also facilitates exhaustion of adoptively transferred engineered T cells, consequently impairing the efficacy of adoptive cell therapy (ACT). Exhaustion is a major mechanism of tumor escape in relapsed cancers following CAR T cell treatment [85], and markers of T cell exhaustion are often associated with poor patient outcomes. In B cell acute lymphoblastic leukemia (B-ALL) patients treated with CD19-targeted, 4-1BB-based CAR T cells, significant DNA methylation reprogramming was observed in CD8+ CD19 CAR T cells four weeks post-infusion, notably methylation of genes associated with effector function and memory potential alongside demethylation of exhaustion-associated transcription factors BATF and thymocyte-selection-associated high mobility group box factor (TOX) [100]. In head and neck cancer, both clinical data and murine models demonstrate that the expression of PD-1 on TILs correlates with clinical outcomes and response to immunotherapy, with tumor rejection in response to anti-PD-1 monoclonal antibody (mAb) therapy associated with a decrease in PD-1high and an increase in PD-1low CD8+ TILs [101]. In chronic lymphocytic leukemia patients treated with CAR T cells, the enrichment and persistence of TIM3+PD-1+ CAR T cells were found to be associated with inferior clinical outcomes [102]. Even before infusion, CAR T cell products that have higher expression of inhibitory receptors, such as PD-1 and LAG-3, exhibit poorer in vivo expansion and antitumor activity in various leukemia and lymphoma patients [103,104].
Apart from cell-surface inhibitory receptors, altered intracellular negative regulators also play a role in T cell dysfunction in cancer. Decreased expression of Src-homology-2-domain-containing tyrosine phosphatase 1 (SHP-1), a cytosolic tyrosine phosphatase, results in hyperactive tyrosine kinase 2 (TYK2) and Janus kinase [105], kinases that facilitate lymphomagenesis [106,107]. In a murine melanoma model, a lower level of phosphorylated Src-homology-2-domain-containing tyrosine phosphatase 2 (SHP-2) was observed and correlated with increased metastasis, heightened release of inflammatory cytokines, and accumulation of myeloid-derived suppressor cells (MDSCs) in the TME [108].
2.1.2. Changes in the Transcriptional and Epigenetic Landscapes of Dysfunctional T Cells and the Implications for T Cell Immunotherapy
Dysfunctional tumor-associated T cells display characteristic changes in their transcriptional landscapes. T cell exhaustion is epigenetically encoded, with exhausted T cells exhibiting reduced chromatin accessibility of T cell effector genes and increased accessibility of genes encoding exhaustion-related transcription factors and inhibitory receptors [85,109,110]. These epigenetic modifications result in various molecular cascades and transcriptional changes that exacerbate the dysfunctional state of TILs. For example, exhausted CD8+ TILs exhibit enriched chromatin accessibility of consensus binding motifs of nuclear factor of activated T cells (NFAT), a transcription factor that directly activates the anergy-inducing proteins gene related to anergy in lymphocytes (GRAIL) and Caspase-3 [47,110,111].
The members of the nuclear receptor subfamily 4A (NR4A1, NR4A2, and NR4A3), all of which are hormone-inducible transcription factors, are additional key regulators in the induction of T cell dysfunction and exhaustion [110,112,113,114,115,116]. Expression of NR4A1, the most extensively studied member of the NR4A family, is triggered by chronic antigen stimulation in solid tumors [115] and remains stable at high levels in tolerant T cells [113]. NR4A1 binds to and inhibits activator protein 1 (AP-1) and nuclear factor-κB (NFκB), thereby repressing IL-2 promoter activation and other cytotoxic effector gene expression [112,113]. NR4A1 further induces T cell dysfunction by promoting H3K27 acetylation and activating tolerance-related genes [113]. CD8+ T cells isolated from cancer patients exhibit high levels of NR4A transcription factors and enrichment of NR4A-binding motifs in accessible chromatin regions [95,109,114,117]. Expression of the NR4A family facilitates exhaustion of not only endogenous CD8+ TILs but also CD8+ CAR+ TILs. This highlights the potential of NR4A as a promising gene-editing target to improve T-cell-based therapies [110,115]. Indeed, studies performed by Chen et al. have demonstrated that triple knockout of NR4A1, NR4A2, and NR4A3 (Nr4a TKO) in CD19-targeted CAR T cells enhances tumor regression in and survival of tumor-bearing mice [115]. Nr4a TKO CAR+ TILs exhibited phenotypic, genotypic, and epigenetic profiles that were similar to those of functional effector CD8+ T cells, further highlighting the significant role that NR4A family members play in T-cell-intrinsic exhaustion [115].
Similar to NR4A1, TOX and TOX2 are key transcription factors highly expressed in TILs that program and maintain exhaustion in both endogenous and adoptively transferred CD8+ T cells [54,55,57,98,118,119,120,121,122]. TOX expression is induced by chronic TCR stimulation and downstream calcineurin and NFAT activation [118,119] and leads to T cell dysfunction by attenuating T cell effector functions and upregulating inhibitory receptors [119]. TOX is highly expressed in TILs, and its expression positively correlates with that of other exhaustion markers, such as PD-1 [118,119]. In HCC, TOX promotes the exhaustion of tumor-antigen-specific CD8+ T cells by inhibiting PD-1 degradation and therefore increasing PD-1 expression on TILs [55,57]. Significantly, TOX is critical for the induction of CD8+ T cell exhaustion; in the absence of TOX, exhausted T cells do not form [54,120]. This critical trait makes TOX another attractive gene-editing target in the development of T-cell-based immunotherapies, a concept actualized by Seo et al. in 2019 [122]. They demonstrated that, compared to wild-type CAR+ TILs, CAR+ TILs with the double knockout of TOX and TOX2 (Tox DKO) exhibited a higher level of effector cytokine production, a decrease in inhibitory receptor expression, and a superior capacity to suppress tumor growth and prolong the survival of tumor-bearing mice [122].
T-box expressed in T cells (T-bet) is a transcription factor that acts as a master regulator of polarization of Th1 T cells, a subset of CD4+ T cells with a high antitumor activity that positively correlates with improved prognosis in patients [123,124,125,126]. T-bet upregulates proinflammatory pathways, directly represses PD-1 expression, and is associated with robust and functional T cells [54,127,128,129]. Decreased levels of T-bet are associated with various malignancies [54,128,129]. In a study by Gacerez and Sentman, overexpression of T-bet in B7H6-targeted CD4+ CAR T cells increased proinflammatory and Th1 cytokine secretion and cellular cytotoxicity against B7H6-expressing tumor cells in vitro, and prolonged survival in an in vivo mice model [124]. These findings suggest that boosting T-bet activity may improve the antitumor efficacy of CAR T cell therapies against solid cancers.
Other exhaustion-related transcription factors, including BLIMP-1, BATF, interferon regulatory factor 4 (IRF4), and Eomesodermin (EOMES), have also been found to be overexpressed in exhausted CD8+ T cells [47,130,131,132]. Interestingly, both increased and decreased levels of BATF and IRF4 in TILs have been linked to oncogenesis [132,133]. Overexpression of BATF and IRF4 has been observed in exhausted T cells and functions to promote exhaustion and repress memory T cell differentiation [132]. By contrast, Seo et al. demonstrated in a murine model that overexpression of BATF and IRF4 in CAR T cells promoted T cell survival, proliferation, memory differentiation, and production of effector cytokines [133]. These contrasting findings may be due to the different models used in the two studies, the first of which analyzed T cell exhaustion in the context of chronic infection, whereas the second focused on the exhaustion of CAR T cells in a tumor mouse model.
T cell signaling and functionality are also impacted by the expression of various phosphatases that act in TCR-initiated signaling pathways. For example, dual specificity phosphatase 2 (DUSP2), a mitogen-activated protein kinase (MAPK) phosphatase that regulates phospho-signaling downstream of the TCR, is highly expressed in tumor-infiltrating CTLs and negatively correlates with cancer patient survival [134]. In terms of its mechanism, DUSP2 recruits the Mi-2β nucleosome-remodeling and histone deacetylase complex, resulting in chromatin remodeling that facilitates T cell exhaustion and dysfunction [135].
Another phosphatase, protein tyrosine phosphatase N2 (PTPN2), has also been linked to T cell dysfunction. PTPN2 contributes to the generation of terminally exhausted CD8+ T cells and negatively impacts αβ TCR signaling. It interferes with cytokine signaling that is essential for normal T cell function, homeostasis, and differentiation by dephosphorylating and inactivating target proteins such as LCK, JAK-1, and JAK-3 from the Janus-activated kinase [105] family, and STAT-1, STAT-3, and STAT-5 from the signal transducer and activator of transcription [136] family [137,138,139,140,141,142]. In a study conducted by Wiede et al. using a mice model, the deletion of PTPN2 in HER-2-specific CAR T cells led to an increase in LCK activation and cytokine-induced STAT-5 signaling, resulting in enhanced CAR T cell activation, homing to, and eradication of target HER-2+ mammary tumors in vivo [137].
Notably, various transcription and epigenetic factors could also play a cooperative role in modulating the dysfunctional T cell phenotype. Through single-cell sequencing of TILs isolated from HCC patients, a variety of transcription factors (e.g., EOMES, T-bet, BLIMP-1, v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog (MAF), TOX, and T cell factor 7 (TCF7)) were shown to regulate CD8+ T cell dysfunction in a complex, interdependent manner [54]. The presence of certain transcription factors, including recombination signal binding protein for immunoglobulin kappa J region (RBPJ), zinc finger BED-domain-containing protein 2 (ZBED2), ETS variant transcription factor 1 (ETV1), inhibitor of DNA binding 3 (ID3), MAF, BLIMP-1, and EOMES, was also determined to be a characteristic of dysfunctional CD8+ T cells in a study conducted with melanoma patients [82]. In lymphomas, the epigenetic modifiers ten-eleven translocation 2 (TET2), DNA methyltransferase 3A (DNMT3A), and isocitrate dehydrogenase 2 (IDH2) are commonly mutated and correlate with heightened hypermethylation of genes involved in TCR signaling and T cell differentiation [75,77,78,143,144,145]. The key altered negative regulators and transcription and epigenetic factors in T cells that contribute to T cell exhaustion and dysfunction are summarized in Table 2.
2.2. Cancer-Cell-Intrinsic Characteristics Hinder Cancer Immunosurveillance and Limit the Efficacy of T-Cell-Based Cancer Immunotherapies
In addition to endogenous T cell dysfunction caused by negative regulator upregulation and changes in the transcriptional and epigenetic landscapes, cancer-cell-intrinsic characteristics also contribute to immune dysfunction and cancer progression (Figure 1).
2.2.1. Molecular Alterations in Tumor Cells
In cancer cells, molecular alterations in key signaling pathways, proto-oncogenes, tumor suppressors, and epigenetics all contribute to cancer development and progression (Table 3).
Key Signaling Pathways Altered in Tumor Cells
Alterations in a myriad of intracellular signaling pathways can contribute to cancer development. In a range of cancer types, genetic alterations in several key signaling pathways have been identified, including the Ras-Erk and PI3K-Akt signaling pathways [222,223,224]. Studies using next-generation sequencing technology conducted with thousands of tissue samples and computational scanning of comprehensive cellular pathway databases have revealed the presence of alterations in additional pathways, such as the Nrf2, Wnt, Myc, p53, Hippo, and Notch pathways [225,226,227,228].
Alterations of Proto-Oncogenes and Tumor Suppressors
Under normal physiological conditions, proto-oncogenes and tumor suppressors act in opposing ways to facilitate homeostatic cell growth [229,230]. Proto-oncogenes stimulate cell division and promote normal cellular growth [230]. Activation via gain-of-function converts proto-oncogenes into oncogenes, which are commonly found in cancer, and results in uncontrolled cell proliferation [230]. Examples of commonly mutated proto-oncogenes include RAS (KRAS, NRAS, and HRAS) and BRAF in the RNA/RAF/MEK/MAPK signaling pathway [230]. Conversion of proto-oncogenes to oncogenes can result from single mutations, such as the common G12V mutation of KRAS, as well as gene amplifications, as seen with c-MYC [231,232,233,234,235,236].
By contrast, tumor suppressors inhibit cell division and prevent cell growth [229]. Inactivation of tumor suppressors via loss-of-function mutations results in a loss of control of cell division and evasion of apoptosis, which contribute to tumorigenesis [229]. A typical and well-studied tumor suppressor is tumor protein p53 (TP53), a transcription factor known as the “guardian of the genome” [237]. TP53 tightly controls cell division and apoptosis, and, in response to DNA damage during cell division, promotes DNA damage repair or apoptotic pathways [237]. Mutations in TP53 can lead to the loss of its tumor suppressor function or the gain of oncogene function [238]. Germline mutations in TP53 cause Li-Fraumeni syndrome [239], a condition in which patients develop early onset breast cancer, sarcomas, or other neoplasms [240].
Common Epigenetic Alterations Identified in Cancer Cells
Epigenetic changes inextricably cooperate with genetic alterations to drive the malignant cancer phenotype. Several factors contribute to epigenetic changes, including DNA methylases/demethylases, histone-modifying enzymes, histone posttranslational modification readers, chromatin/nucleosome remodelers, and microRNA (miRNA) [241,242]. Hypermethylation in the promoter region of tumor suppressors is commonly seen in cancers. For example, hypermethylation of retinoblastoma protein (RB), phosphatase and tensin homolog (PTEN), breast cancer type 1 susceptibility protein (BRCA1), breast cancer type 2 susceptibility protein (BRCA2), O6-methylguanine-DNA methyltransferase (MGMT), cyclin-dependent kinase inhibitor 2B (CDKN2B), and Ras-association-domain-containing protein 1A (RASSF1A) are all reported in cancer, and result in the loss of ability to protect against tumorigenesis [243,244,245,246]. Moreover, histone modifications mediated by histone acetyltransferases (HATs), deacetylases (HDACs), methyltransferases (HMTs), and demethylases (HDMs) at regulatory regions can also lead to aberrant gene expression via the introduction or removal of acetyl or methyl groups, respectively [247,248,249]. For instance, the removal of acetyl groups from the promoter of the CDKN1A gene by HDACs leads to its downregulation and is linked to the onset of various cancers [202]. Finally, alterations in chromatin remodeling complexes are also responsible for the development of cancer. Changes in the switch/sucrose non-fermenting (SWI/SNF) complex are often observed in melanomas and renal carcinomas and correlate with poor survival rates [250]. Mutations in the AT-rich interactive-domain-containing protein 1A (ARID1A) gene, which encodes BAF250a, a component of the SWI/SNF family of chromatin remodeling complexes, are frequently observed in ovarian clear cell carcinomas, endometrioid carcinomas, pancreatic adenocarcinomas, transitional cell carcinomas, and triple-negative breast cancers [208,209,251].
2.2.2. Immunosuppressive Tumor Microenvironment
Metabolic Competition in the TME
Similarities can be observed between the metabolism of cancer cells and activated T cells due to their shared characteristics of rapid growth and proliferation [252]. While most cell types prioritize oxidative phosphorylation as the primary energy production pathway in the presence of oxygen, both cancer cells and activated lymphocytes exhibit “aerobic glycolysis” as their main metabolic program, whereby they preferentially utilize glycolysis for energy production even when sufficient oxygen is available to support oxidative phosphorylation [252,253]. This preference for glycolysis over oxidative phosphorylation is driven by the need for rapid biosynthesis, which glycolysis can efficiently achieve, whereas oxidative phosphorylation emphasizes efficient ATP production [252]. This similarity in metabolic profiles between cancer cells and T cells is the foundation of metabolic competition in the TME [254]. The metabolic restriction of T cells caused by tumor glucose consumption leads to decreased mTOR activity, glycolytic capacity, and IFN-γ production in T cells, ultimately facilitating T cell dysfunction and enabling tumor progression [254].
Nutrients and Metabolites
Cancer cells characteristically exhibit an elevated glycolysis rate, which consequently depletes the TME of the glucose required for normal T cell effector function and hence impairs the immune response against cancer [254,255]. In some cancers, such as squamous cell carcinoma and pancreatic adenocarcinoma, this increase in glycolysis is facilitated by the overexpression of glucose transporter 1 (GLUT-1) [256,257]. The high glucose uptake and glycolysis rate exhibited by cancer cells correlate with lower CD8+ T cell infiltration and poorer patient survival [256,257]. In terms of the effect on T cells, a lack of glucose hinders T cell effector functions by inhibiting mammalian target of rapamycin complex 1 (mTORC1) activation and downstream Myc induction, impairing NFAT signaling, and suppressing IFNγ translation [258,259]. Significantly, glucose depletion does not impair the differentiation or function of Tregs; in fact, glucose-deficient environments promote Treg induction and activity in vitro [260]. Collectively, these observations demonstrate that tumor-cell-mediated glucose depletion enhances the immunosuppressive character of the TME by categorically favoring Treg survival and function [255,260].
Cells also compete for glutamine in the TME, an essential nutrient for tricarboxylic acid (TCA) cycle maintenance and lipid synthesis in both cancer cells and effector T cells [261,262,263]. In various cancers, alanine, serine, and cysteine transporter 2 (ASCT2), the major glutamine transporter, are overexpressed, giving these cancer cells an advantage over other cells in glutamine uptake [264].
Arginine is an essential nutrient for the urea cycle and polyamine synthesis and is required by both cancer and T cells [261]. A variety of cancer cells overexpress nitric oxidase synthase and arginase, major enzymes in arginine metabolism, therefore allowing them to outcompete other cell types for arginine uptake [265,266]. Evidence from mouse models suggests that increasing the L-arginine concentration can result in a shift in T cell metabolism from glycolysis to oxidative phosphorylation, promoting the generation of central-memory-like T cells with enhanced antitumor activities and longer persistence [267].
Indoleamine-2,3-dioxygenase (IDO), an enzyme that converts tryptophan to kynurenine, has elevated activity in various cancers, resulting in a depletion of the essential nutrient tryptophan and an accumulation of by-product kynurenine in the TME [268,269,270]. Tryptophan deficiency impairs T cell effector function by inhibiting mTORC1 activity and downregulating TCR-CD3-ζ [271]. Furthermore, elevated IDO activity increases the level of kynurenine in the TME, and this metabolite impairs TCR signaling and therefore facilitates T cell dysfunction [255].
Tumor growth can lead to a decrease in oxygen perfusion and result in a hypoxic TME, a signature of advanced cancer [255]. Hypoxic environments promote tumor progression by facilitating CD8+ T cell dysfunction via various mechanisms [272]. Hypoxia directly impacts T cells by inducing them to differentiate into FoxP3+ Treg cells, which, under hypoxic conditions, produce extracellular adenosine that further suppresses T cell effector function [272,273,274,275]. Additionally, exposure to hypoxia in the TME increases histone bivalency in CD8+ TILs, leading to a reduction in their transcriptional potential and contributing to the development of T cell exhaustion [276]. Hypoxia also directly contributes to T cell exhaustion by driving T cell expression of CD39, an ectoenzyme that secretes immunosuppressive adenosine [277]. Furthermore, hypoxia also indirectly hinders antitumor immunity by impacting tumor-associated immune and stromal cells. Under hypoxic conditions, tumor cells and associated cells secrete cytokines and factors that attract Tregs to the TME [278]. Hypoxic conditions within the TME also stimulate the expression of hypoxia-inducible factors (HIFs) in tumor cells [272,279,280]. HIF-1α induces the expression of vasculature endothelial growth factor (VEGF), a pro-angiogenic factor that promotes the formation of abnormal tumor vasculature characterized by a deficiency in pericytes and a leaky basement membrane [272,279,280]. Consequently, this prevents lymphocyte extravasation from blood vessels into the TME and facilitates immune exclusion [272,279,280]. HIF-1α also stimulates the expression of the immune checkpoint inhibitors PD-L1, CTLA-4, and LAG-3 on tumor and tumor-associated immune and stromal cells [272,279,280]. In the TME, low oxygen levels also inhibit T cell proliferation by affecting potassium concentration, although the exact mechanism is unknown. Hypoxia has been shown to reduce the expression of Kv1.3 potassium channels in T cells, which play a crucial role in maintaining the membrane potential and function of T cells [281]. However, another hypothesis suggests that necrotic tissue in the TME contributes to high potassium concentration in the extracellular fluid, resulting in AKT/mTOR hypophosphorylation downstream of the TCR and significantly suppressing T cell effector function [282]. This presents a paradoxical contrast to the first hypothesis. Baixauli and Vodnala found that a high K+ concentration can trigger stem-cell-like characteristics in TILs, rendering them more effective in mediating tumor destruction following immune checkpoint inhibitor therapy [283,284]. Accordingly, highly proliferative TILs, characterized by elevated expression of the proliferation marker Ki67, also express high levels of Kv1.3 channels [285].
Calcium (Ca2+) is also critical to various cellular functions, including signaling, metabolism, apoptosis, and cell division [286,287,288]. Increased calcium levels in cancer cells confer a growth advantage, impairing calcium uptake by T cells in the TME and consequently hindering T cell activation and proliferation [289,290,291]. Using targeted optogenetic stimulation of intracellular Ca2+ signaling, Kim et al. successfully restored the cytotoxic effector functions of T cells in tumor sites [292].
TME-Derived Metabolites
Reactive oxygen species (ROS) are mainly generated as a by-product of energy production and are known to be markers of cell stress and inducers of cellular damage and death. ROS also affect T cell metabolism and are therefore key modulators of the antitumor activity of T cells. Sustained high levels of ROS are linked to immunosuppression in the TME [293,294]. They can impede T cell function in a multitude of ways, including inhibition of the DNA-binding capacities of NFAT and NKκB [295] and disruption of pMHC–TCR binding [296]. In the TME, various tumor-associated immune and stromal cells contribute to high levels of ROS. For example, immature myeloid cells, myeloid-derived suppressor cells, and activated granulocytes produce ROS, which results in apoptosis and suppression of T cells [294,296,297,298]. Interestingly, while high levels of ROS are detrimental to T cells, moderate levels of ROS are required for normal TCR signaling and T cell activation [293,294,295].
Elevated levels of cholesterol within tumors also contribute to the immunosuppressive environment of the TME. High cholesterol increases endoplasmic reticulum (ER) stress in cytotoxic T cells and thereby induces upregulation of inhibitory receptors in T cells, including PD-1, 2B4, TIM-3, and LAG-3 [299].
Additionally, excessive secretion and subsequent accumulation of lactic acid by tumor cells lowers the pH of the TME. The acidic environment negatively affects T cell effector function by suppressing the PI3K/Akt/mTORC1 pathway and glycolysis, pathways that are critical for normal T cell activation, proliferation, and function [300,301,302]. However, Tregs have a metabolic advantage over conventional T cells due to the action of the Treg transcription factor FoxP3, which reprograms Treg metabolism to better adapt to the lactic-acid-rich TME. FoxP3 suppresses Myc and glycolysis, enhances oxidative phosphorylation, and increases nicotinamide adenine dinucleotide (NAD) oxidation, optimizing the metabolic profile of Tregs to suit the low-glucose, high-lactate TME [303].
Tumor-Associated Immune and Stromal Cells
Various resident or recruited immune and stromal cells within the TME significantly contribute to the immunosuppressive environment. The dual influence of these cells on both cancer progression and effector T cell dysfunction is illustrated in Figure 2.
- Tumor-Associated Macrophages (TAMs)
Tumor-associated macrophages (TAMs), infiltrating macrophages characterized by a pro-tumorigenic M2-like phenotype, are present in solid tumors and critically contribute to the pro-tumoral environment of the TME by facilitating tumor growth, angiogenesis, and metastasis while attenuating antitumor immune responses [304,305,306,307]. TAMs accumulate and are retained in the hypoxic TME due to the presence of macrophage chemoattractants (e.g., monocyte chemoattractant protein (MCP)-1 and macrophage inflammatory protein (MIP)-1α) [308,309]. TAMs advance tumor initiation, progression, and metastasis by secreting immunosuppressive cytokines, chemokines, proteolytic enzymes, and growth factors and by upregulating immune checkpoints on T cells [307]. IL-6, IL-23, and IL-17 secreted by TAMs promote inflammation within the TME that drives tumor growth [310,311]. TAMs further facilitate metastasis by promoting epithelial–mesenchymal transition (EMT) via the secretion of IL-1β, IL-8, TNF-α, and TGF-β [312,313,314]. Matrix metalloproteinases (MMPs), cathepsins, and serine proteases produced by TAMs mediate tumor-cell–ECM interactions and ECM degradation, thereby aiding the intravasation of tumor cells [315,316,317]. TAMs also play a role in tumor angiogenesis and the remodeling of existing vasculature via the production of factors such as MMP-9, VEGF, FGF-2, CXCL8, IL-1, IL-18, COX-2, iNOS, and MMP7 [318,319,320,321,322]. In addition to facilitating the progression and metastasis of cancer cells within the TME, TAMs also promote the survival of circulating tumor cells (CTCs) by activating the PI3K/Akt survival signaling pathway in cancer cells and secreting protective chemokines and cytokines [323,324,325]. In clinical studies, a high TAM count has been found to correlate with a poorer prognosis in patients with various cancer types [136,326,327,328].
TAMs facilitate CD8+ T cell dysfunction via several mechanisms. TAM-derived secretory factors, including ARG1, iNOS, TGF-β, IL-10, and ROS, induce CD8+ T cell exhaustion and dysfunction [329,330]. TGF-β elevates the expression of the checkpoint inhibitory molecules TIM-3, PD-1, and CTLA-3 on T cells and inhibits IFNγ and granzyme B secretion [331]. Furthermore, TAMs express PD-L1 and induce its expression on monocytes, which promotes T cell exhaustion upon ligation [330,332]. TAMs further attenuate effector T cell function by secreting high levels of IDO and depleting the TME of essential amino acids, including L-arginine and tryptophan [332].
- Cancer-Associated Fibroblasts (CAFs)
Various signaling molecules within the TME, including TGF-β, ROS, inflammatory cytokines, contact signals, and receptor tyrosine kinase (RTK) ligands, trigger the conversion of fibroblasts into activated cancer-associated fibroblasts (CAFs) that exhibit significant heterogeneity and plasticity [333,334]. CAFs affect the production and structure of the ECM and secrete factors that promote tumor growth and suppress antitumoral immunity. CAFs deposit and remodel the ECM by secreting matrix-crosslinking enzymes, which contribute to the structural stiffness of tumor tissue [333,335,336,337]. The rigidity of tumor tissue promotes cancer growth by triggering survival and proliferation signaling within cancer cells and impedes immune cell infiltration and function by collapsing vasculature and creating a hypoxic environment [337,338,339,340]. Interestingly, CAFs also induce angiogenesis in a manner that selectively promotes tumor cell growth [341]. CAF-derived matrix proteases mediate tumor matrix remodeling that excludes CD8+ T cell infiltration, promotes local cancer cell invasion, and increases the metastatic potential of cancer cells [341,342,343,344,345]. Several matrix components, such as tenascin and periostin, also enhance cancer cell survival by increasing Wnt signaling [346,347]. Tumor growth and survival are further supported by CAF-derived growth factors, cytokines, and exosomes, including TGF-β, LIF, GAS6, FGF5, GDF15, and HGF, which collectively act to promote the invasive and proliferative potential of cancer cells [348,349,350,351,352,353,354].
CAFs indirectly facilitate T cell dysfunction by secreting CXCL1 and CXCL2, which polarize macrophage differentiation into generating the immunosuppressive M2 and TAM subtypes [332]. Ligation of the CAF inhibitory ligands PD-L2 and FasL to their corresponding receptors on CD8+ T cells directly induces effector T cell dysfunction [332]. The cytokines and chemokines secreted by CAFs also influence a range of immune cells, resulting in both immunosuppressive and immunoenhancing outcomes [341]. Although CAFs play a dual role in immune response regulation, they predominantly create an immunosuppressive environment. For example, IL-6, CXCL9, and TGF-β secreted by CAFs attenuate T cell effector responses [333,355]. CAFs have also been shown to engage in antigen cross-presentation that activates CD4+ T cells and suppresses CD8+ T cells, an observation further supported by clinical studies [356,357,358]. This effector T cell suppression is mediated by PD-L1 and PD-L2 expression on CAFs, which inhibits the production of IL-2, a major T cell growth factor [359].
- Tumor-Associated Neutrophils (TANs)
Tumor-associated neutrophils (TANs) are another class of cells closely associated with the TME. Like macrophages, TANs can polarize toward an anti-tumoral subtype (N1 TANs) or a pro-tumoral subtype (N2 TANs) [360,361,362,363,364]. N2 polarization is stimulated by the presence of TGF-β in the TME [361,365]. While TANs have been shown to exhibit both pro-tumoral and anti-tumoral functions, this section focuses on their pro-tumoral functions, including the promotion of angiogenesis, EMT transition, migration and invasion, and immunosuppression [366]. It is important to note that regarding the functions of TANs, contradictory evidence currently exists in the literature due to the high heterogeneity of the neutrophil population within the TME and insufficient markers being available to distinguish different subtypes [366]. Furthermore, a reliance on murine models has resulted in conclusions being drawn that are not necessarily relevant in clinical settings [367].
TANs facilitate tumor initiation via the actions of neutrophil elastase (NE), ROS, and reactive nitrogen species (RNS) [294,368,369,370]. They release and/or induce the production of genotoxic substances (e.g., ROS) that damage the DNA of epithelial cells and facilitate carcinogenesis [371]. Various proteases secreted by TANs, including NE and MMP9, activate growth factor pathways within cancer cells and enhance hyperproliferation [371,372]. TANs also generate neutrophil extracellular traps (NETs) composed of DNA–histone complex, proteins, and fibers secreted by activated neutrophils, and these facilitate tumor progression and metastasis by sequestering and promoting the adhesion of circulating cancer cells in distant organs [373,374,375]. TANs also play a protective role in the survival of CTCs. CTCs associated with neutrophils have been found to demonstrate greater proliferation and enrichment of positive regulators of cell cycle progression compared to CTCs alone [376]. Furthermore, TAN-derived MMP9 and VEGF facilitate angiogenesis [377,378]. TANs also trigger EMT via the NE cleavage of E-cadherin, upregulation of EMT-related genes (including TWIST and Vimentin), facilitation of nuclear translocation of β-catenin, and promotion of nuclear expression of ZEB1, a transcription factor that boosts tumor invasion and metastasis [379,380,381].
TAN-derived cytokines and chemokines also interact with other immune cells within the TME to promote an immunosuppressive environment. TANs suppress T-cell-mediated immunity by releasing high levels of arginase and protumor factors, including CCL2, CCL5, NE, and cathepsin G [377,382]. Additionally, TAN-derived CCL17 recruits Tregs to the TME, while CXCL14 attracts M2 macrophages [383]. ROS, H2O2, and ARG-1 produced by TANs further contribute to immunosuppression [361,384].
3. Exploring the Therapeutic Landscape for Cancer Treatment
Many traditional cancer treatments, such as chemotherapy and radiotherapy, have severe side effects and fail to specifically target cancer cells. Since the accomplishment of the human genome project, progress in the development of high-throughput, low-cost technologies has enabled significant advancement in the generation of novel cancer treatments, better prediction of patient outcomes, and the development of targeted therapies. A summary of the traditional therapies and the novel targeted approaches used in cancer treatment is shown in Figure 3.
3.1. Targeted Therapies: Small-Molecule Drugs, Therapeutic Antibodies, Oncolytic Viruses, and Gene Editing
Targeted therapies are a newer approach to cancer treatment designed to selectively destroy cancer cells, sparing normal cells and reducing toxic side effects [385]. More than 80 anticancer small-molecule drugs have been approved by the US FDA [386]. For example, various small-molecule drugs have been developed that successfully target the G12C mutation commonly seen in KRAS, a mutation previously deemed impossible to target due to its structural and biochemical properties [387,388,389,390].
Since the development of hybridoma technology in 1975 by Milstein and Köhler, therapeutic antibodies based on monoclonal antibodies (mAbs) or polyclonal antibodies (pAbs) have evolved and undergone a revolution [391]. Unlike small-molecule drugs that penetrate the cell membrane to block signaling, antibodies mainly bind to receptors or ligands on the cell surface and have various therapeutic mechanisms of action. For instance, antibodies can block the binding of signaling molecules, thereby inhibiting cell proliferation or facilitating cell death through the inhibition of DNA repair, prevention of cell cycle progression, and/or induction of apoptosis [392,393,394,395]. Therapeutic antibodies can also recruit immune effector cells to trigger antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC) [396].
Another promising new therapy is oncolytic virus therapy. Due to the lack of functional antiviral mechanisms in cancer cells, oncolytic viruses can be utilized to selectively replicate in and lyse cancer cells while sparing normal tissue [397,398]. The concept was initiated in the early 1900s, and in 2015, T-VEC became the first FDA-approved vaccine for the treatment of melanoma [399].
Gene-editing technologies are also being utilized in targeted therapeutic approaches to correct mutations in both cancer cells and T cells, thereby strengthening the antitumor response. The clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 system is a widely used gene-editing tool known for its simplicity, broad applicability, and high potential [400]. In recognition of its impact, the CRISPR/Cas9 system was awarded the Nobel Prize in Chemistry in 2020 [400]. The CRISPR/Cas9 system is rapidly gaining popularity in the field of cancer immunotherapy. For instance, it can be utilized to correct mutations in the TP53 gene, thereby restoring its tumor-suppressing properties and reducing the growth of tumors [401]. Additionally, the use of multiple sgRNAs to target the human estrogen receptor 2 (HER2) gene has been shown to effectively decrease HER2 expression and impede the growth of HER2-positive cancer cells [402]. In addition to its application in editing sites within tumor cells, CRISPR/Cas9 technology has been widely utilized to restore and enhance T cell function. For example, CAR T cells that have been engineered to lack PD-1 or CTLA-4 exhibit increased cytotoxicity, robust cytokine release, and a potent antitumor response [403,404]. By targeting the integration of the CAR construct into the TCR α constant region, universal expression of CAR and prolonged T cell persistence in vivo can be achieved [405,406]. The technology can also be used to generate universal “off-the-shelf” CAR T cells by eliminating endogenous TCR, MHC, and CD52 molecules [407]. Furthermore, mutations in T cell signaling can be corrected to restore normal T cell function [408].
3.2. Significant Emerging Immunotherapies: Cancer Vaccines, Engineered T Cells, and Immune Checkpoint Inhibitors
Of the various advancements in cancer treatment over the past decade, immunotherapy stands out as a particularly promising approach. Its remarkable efficacy and specificity earned it the title of Science Breakthrough of the Year in 2013 [409]. Immunotherapy is a targeted cancer treatment that utilizes a patient’s own immune system to eliminate cancer cells [409]. Its effectiveness lies in its ability to target neoantigens, unique antigens generated by tumor-specific mutations [410]. By doing so, the immune system can differentiate between cancer and non-cancer cells, allowing it to specifically attack and destroy the former while leaving healthy tissue unharmed [410]. Several categories of cancer immunotherapies exist, including cancer vaccines, CAR T cells, TCR T cells, and TILs, all of which have demonstrated durable clinical responses.
In the past decade, several anti-neoantigen cancer vaccines have been developed [411]. The first cancer vaccine consisted of dendritic cells loaded with neoantigens and was successful in eliciting specific T cell responses in melanoma patients [412]. Sahin et al. then proved the feasibility of “individualized mutanome vaccines”—vaccines tailored to the mutations in each individual cancer patient—paving the way for personalized cancer immunotherapy [413]. Vaccines containing synthetic long peptides designed using bioinformatics have also been shown to improve the overall survival of cancer patients [414,415]. mRNA vaccines, known for their versatility in design and target selection, have also been widely used in cancer treatment and prevention [416]. For example, Rosenberg and colleagues developed an mRNA vaccine for gastrointestinal cancer patients that elicited neoantigen-specific T cell immunity [417]. This was demonstrated by the successful isolation and verification of T cells targeting KRAS G12D from treated patients, demonstrating the safety and efficacy of mRNA vaccines [417].
Furthermore, the incorporation of heat shock protein (HSP)-based components into cancer vaccines has been shown to improve treatment efficacy [418]. Heat shock proteins (HSPs), whose expression is induced by heat shock or other cellular stressors, are a large family of proteins involved in protein folding and trafficking that also play a pathological role in cellular proliferation, differentiation, and carcinogenesis [418]. Because HSPs are significantly overexpressed in a wide range of human cancers and have the promiscuous ability to chaperone and present a broad repertoire of tumor antigens to APCs, they are used in cancer immunotherapy as a means of promoting cancer antigen-specific T cell stimulation [419,420,421]. When cancer cells undergo stress, such as during chemotherapy or radiation therapy, they release HSPs into the extracellular space [422]. These extracellular HSPs, in complex with tumor-associated antigens, can then bind to receptors expressed on APCs, leading to endocytosis, antigen cross-presentation to MHC I molecules, T cell priming, and induction of antigen-specific CTLs [422,423,424]. Cancer vaccines incorporating HSP–peptide complexes have demonstrated successful elicitation of immune responses directed specifically against the tumor from which the HSPs were isolated [422,425,426,427].
The development of therapeutic antibodies has greatly contributed to the advancement of various immunotherapies. Antibodies that mimic TCR function have been successfully developed due to the high efficiency with which peptide–MHC complexes can be purified and refolded in vitro [428,429,430,431,432,433]. The versatility of antibodies has enabled their modification for different purposes, expanding their use in cancer immunotherapy, such as in the design of bi-specific T cell engagers (BiTE), antibody–drug conjugates (ADC), and CAR T cells [431,434,435,436,437,438,439,440]. Immune checkpoints, such as CTLA-4 and PD-1, play a role in negatively regulating the immune response. Antibody therapies that target these checkpoints and their ligands have been shown to produce durable and persistent clinical responses in various cancer patients through the restoration of T cell function [441].
3.3. Challenges and Emerging Strategies in Cancer Immunotherapy
3.3.1. Combating Antigen Escape in CAR T Cell Therapy
Despite the progress made in cancer immunotherapy, various obstacles still need to be addressed. For example, antigen escape due to the loss or mutation of the target antigen is commonly seen in relapsed patients treated with CD19-targeting CAR T cell therapy [442]. Identifying target antigens that are consistently and exclusively expressed above a detectable threshold on tumor cells, while not being expressed on essential healthy cells, is a critical challenge in developing safe and efficacious CAR T cell therapies for solid tumors [443]. To combat the challenge posed by antigen escape, various pharmaceutical strategies have been developed that can selectively upregulate the expression of cell-surface target antigens on cancer cells, thereby increasing target antigen density recognition by CARs [443]. For example, the use of epigenetic modulators as sensitizers for CAR T cell therapy has been successfully demonstrated in multiple preclinical studies. Anurathapan and colleagues established that exposure to decitabine, a DNA methyltransferase inhibitor, upregulates cell-surface expression of mucin short variant S1 (MUC1) on pancreatic cancer cells, thus making tumor cells more susceptible to in vitro cytolysis by MUC1-specific CAR T cells [444]. Besides epigenetic modulators, other compounds, including protein kinase C modulators [445] and γ-secretase inhibitors [446], have been demonstrated to successfully upregulate antigen expression on target cells and abate immune escape of tumor cells expressing low levels of the target antigen.
Multiple Antigen-Targeting Strategies in CAR T Design
While the upregulation of a single target antigen can increase the efficacy of CAR T cell therapy against certain malignancies, the highly heterogenous nature of most cancers renders single-antigen-targeting strategies suboptimal in many cases. To tackle the challenge of phenotypic heterogeneity, various innovative CAR T therapies designed to target multiple antigens are being developed.
Combination of Single-Antigen Specific CAR T Therapies
The most straightforward method entails the co-administration of two single, specific CAR T cell products, such as both CD19-targeted and CD22-targeted CAR T cells, either sequentially [447] or simultaneously [448]. Bispecific CAR T cells have also been developed, which coexpress two different CAR transgenes and are activated by recognition of either antigen A or antigen B [449].
Tandem CARs
Another design that allows multiple antigen targeting involves tandem CAR T cells, which combine the antigen-recognition scFvs for two distinct antigens in a manner so that the combined exodomain can co-engage both antigens together in a bivalent immune synapse [450,451,452]. Tandem CARs exhibit T cell activation upon recognition of either antigen, and binding to both targets results in an even more potent T cell response [443].
Combination of CAR T Cells and BiTEs
In another innovative method, CAR T cell targeting is combined with the release of bispecific T cell engagers (BiTEs), fusion proteins that consist of two scFvs—one that binds to T cells via the CD3 receptor, and the other that recognizes a tumor-specific antigen [453]. Upon activation, these CAR T cells synthesize and release BiTEs, which subsequently recruit bystander T cells to elicit an immune response against a second tumor-associated surface antigen [454].
Modular CAR T Designs
Finally, the invention of modular CAR T designs has greatly increased the flexibility and ease of design, enabling the generation of a large panel of CARs against numerous tumor-associated antigens [455,456,457]. In this approach, T cells are genetically modified to express a ‘universal’ CAR that targets a non-human molecule, such as biotin or fluorescein isothiocyanate (FITC), instead of a tumor-associated antigen [443]. Subsequently, these CAR T cells can be activated by administering bispecific adapters, such as an FITC-labeled tumor antigen, that cross-link the CAR T cells to the specified antigen [443]. The modular design offers a significant benefit by enabling the sequential or simultaneous infusion of various adapters to target a broad range of heterogeneously expressed antigens [443,456].
3.3.2. Addressing Additional Challenges: T Cell Trafficking and Persistence
Another challenge to overcome is poor T cell trafficking, which contributes to the lack of success of CAR T cells against solid tumors, despite its remarkable response rates against hematological malignancies. To ameliorate T cell trafficking to and penetration of the tumor site, one straightforward solution is to perform local administration of T cells directly into the tumor site, rather than systemically via intravenous injection [458]. Another approach used to improve T cell trafficking involves engineering T cells to express chemokine receptors that match the chemokines present in the TME [459]. For example, CAR T cells engineered to overexpress CXCR1 or CXCR2 demonstrated significantly enhanced T cell trafficking and antitumor responses [460,461]. Additional chemokine receptors, including CCR2, CCR5, CCR6, and CXCR3, are known to be used for effector T cell recruitment to the tumor site, indicating their potential for use in therapeutic applications [462,463].
A lack of long-term persistence following the adoptive transfer of CAR T therapies presents another significant barrier in the development of effective CAR T therapies against solid cancers. A plethora of factors have been shown to contribute to the poor long-term persistence of adoptively transferred T cells, including patient preconditioning, ex vivo culture conditions, development of T cell exhaustion, design of the CAR construct, and host immune response against the infused product [464,465,466,467,468,469]. In order to achieve long-term persistence, adoptively transferred T cells must infiltrate secondary lymphoid organs to continually renew the population of circulating T cells that subsequently migrate to the tumor sites [470,471]. A method to improve persistence was elucidated by Xu and colleagues, who successfully demonstrated that the addition of IL-7 and IL-15 in cell culture media during ex vivo expansion of CAR T cells increased the frequency of CD8+ T cells positive for CD45RA, a marker of naïve T cells, and CCR7+, a chemokine receptor essential for T cell homing to secondary lymphoid organs [470]. This subset of CD8+CD45RA+CCR7+ T cells exhibited robust antitumor activity even with repetitive antigen challenges and maintained migration to secondary lymphoid organs, therefore achieving prolonged persistence [470].
Furthermore, solid cancers present the additional challenge of tumor-type-specific mechanisms that hinder the therapeutic efficiencies of CAR T therapies. Different solid cancers have distinct characteristics, such as an exceptionally dense tumor stroma, heterogenous expression of cancer antigens, or lack of strong immunogenic neoantigens—factors that deter the ability of CAR T cells to infiltrate the tumor and recognize and eliminate cancer cells [472,473]. For example, pancreatic ductal adenocarcinoma (PDAC) is characterized by an extremely dense stroma deposition surrounding legions that presents a physical barrier against immune infiltration [474]. In glioblastoma, profound tumor-mediated immune suppression and the blood–brain barrier pose significant challenges to the penetration and function of therapeutic immune cells [475]. In preclinical studies aiming to treat pancreatic and prostate cancer using CAR T cells targeting mucin-1 (MUC1) and prostate stem cell antigen (PSCA), low expression of the target antigens on the tumor cells led to tumor escape and a failure of the CAR T cells to eradicate the tumors [444]. Similarly, insufficient anaplastic lymphoma kinase (ALK) target density on neuroblastoma cells limited the therapeutic efficacy of ALK-specific CAR T cells [476]. Overall, various tumor-specific factors can collectively contribute to the limited efficacy of CAR T cell therapies against solid tumors, emphasizing the need for continued research efforts to develop effective strategies to overcome these barriers.
3.3.3. Combinatorial Strategies for CAR T Therapy
In recent years, combination therapy has emerged as a significant method to overcome the obstacles faced by immunotherapy [477]. Combinatorial strategies include administering T cell therapies along with traditional therapies such as chemotherapy [478] and radiotherapy [479], or with other targeted therapies and immunotherapies [477].
For example, the use of small molecules to blockade the PI3K-Akt-mTOR pathway is shown to enhance in vivo expansion and functionality of CAR T cells by promoting the development of T memory stem cell (Tscm), central memory T cell (Tcm), and naïve T cell (Tn) populations, decreasing exhaustion marker expression, and increasing the ratio of CD8/CD4 T cells [477,480,481]. In patients with relapsed or refractory multiple melanomas (MMs), combination therapy with BCMA-targeted CAR T cells along with γ-secretase inhibitor (GSI), a small molecule inhibitor that effectively prevents γ-secretase-mediated cleavage of BCMA on MM cells, successfully increased BCMA surface expression on tumor cells and improved patient response rates [446].
T-cell-based therapies can also be combined with other immunotherapies to enhance their effectiveness. For instance, lenalidomide is an immunomodulatory drug that activates NK cells, heightens immune surveillance, and exhibits tumoricidal effects on MM [482]. When combined with CAR T therapy, lenalidomide enhances CAR T cell function by increasing cytotoxicity, enhancing the maintenance of memory T cell populations, and promoting the formation of immune synapses [477,483,484].
The combination of CAR T cell therapy with immune checkpoint inhibitors is also a field of great interest. Notably, the administration of monoclonal antibodies against PD-1/PD-L1, such as pembrolizumab, nivolumab, and atezolizumab, has improved the therapeutic efficiency of CAR T cell therapy for certain patient populations [477,485,486]. The combination of CAR T therapy with other immune checkpoint inhibitors, such as monoclonal antibodies against CTLA-4 (NCT00586391) and TIGIT [477] is also being explored. The use of combination therapy has emerged as a key strategy to tackle various obstacles faced in the field of immunotherapy. Extensive investigation into novel combinational strategies is urgently needed to achieve greater clinical therapeutic success.
3.3.4. Multidimensional Omics Data Analyses in the Advancement of T Cell Immunotherapy
Recent advances in omics technologies have generated a vast amount of data and information. Analysis of multidimensional data encompassing genomics, epigenomics, transcriptomics, T cell receptor repertoire profiling, proteomics, metabolomics, and microbiomics is a powerful approach and can be the key to overcoming the current obstacles associated with T cell immunotherapy [487]. Indeed, the integration of multi-omic data analyses provides researchers with the ability to more accurately identify targets for the development of multiple-antigen-targeting CAR T cells that can minimize on-target, off-tumor toxicities, antigen escape, and therapy resistance [487,488,489,490,491]. For instance, combining multiple CAR targets utilizing Boolean “AND” and/or “AND-NOT” logic gating improves tumor-targeting specificity and reduces toxicity [492,493], an approach made possible through the use of comparative analyses of genomics, transcriptomics, and proteomics data from both tumor and non-malignant tissue samples to identify ideal targets [489,491].
Moreover, the multidimensional data generated by omics analyses can help researchers identify the expression of specific transcription factors, distinct epigenetic signatures, and unique metabolic states that are correlated with the enhanced therapeutic effectiveness of CAR T cells, therefore providing valuable insights into the underlying mechanisms driving CAR T cell efficacy and persistence [133,487,494,495,496]. Multi-omics data can also be used to identify tumor cell characteristics that influence response to CAR T cell therapy, features of the tumor microenvironment that can hinder CAR T cell efficacy, and potential associations between CAR T cell efficacy and the gut microbiota and microbial metabolites [487]. Undoubtedly, the leveraging of multi-omics data analyses significantly facilitates the development of novel methods to improve CAR T therapy, making it an increasingly significant research methodology.
3.3.5. Advanced Methods for Monitoring CAR T Cell Potency in Patients: Flow Cytometry, CyTOF, and ImmunoPET
Non-invasive, longitudinal monitoring of CAR T cell kinetics in patients following CAR T cell infusion can provide important insights into the efficacy and activity of the therapy. Various flow cytometry assays are one of the most common methods to monitor CAR T cell populations in patients. For example, Blache and colleagues developed a 13-color/15-parameter flow cytometry panel that can characterize immune cell subpopulations in patients’ peripheral blood during CAR T cell treatment [497].
A novel approach for monitoring CAR T cell potency involves the application of cytometry by time of flight (CyTOF), which can be used to analyze trafficking and functional protein expression in CD19 CAR T cells across various patient samples and correlate them with T cell phenotypes [498]. Comparison between non-engineered T cells derived from leukapheresis, CAR T cells pre-infusion, and CAR T cells post-infection derived from peripheral blood, bone marrow, or cerebrospinal fluid of patients, revealed disparate expression profiles, indicating the spatiotemporal plasticity of CAR T cells in patients [498].
Another method for monitoring the potency of CAR T cells in patients is immune positron emission tomography (immunoPET), an imaging technique that combines the targeting specificity of mAbs with the high sensitivity of PET, enabling the visualization of the in vivo dynamics of CAR T cells [499]. ImmunoPET was successfully applied to noninvasively visualize CAR T cells by Simonetta and colleagues, who identified inducible co-stimulator (ICOS), a molecule primarily expressed on activated T cells, as highly expressed in CAR+ T cells compared to non-genetically modified CAR– T cells [500]. This expression disparity between CAR+ and CAR– T cells in patients underlined the suitability of ICOS as a target for immunoPET [499].
4. Conclusions
In recent years, the advent of immunotherapy has marked a paradigm shift in the treatment of cancer. This innovative approach has facilitated the development of a range of cutting-edge immunotherapies that have significantly improved the longevity of cancer patients. However, despite the promising advances and growing success of cancer immunotherapy, challenges still exist in the field. One such challenge is antigen escape due to the loss or mutation of the target antigen, which can be addressed through the use of pharmaceutical strategies that upregulate the expression of cell-surface target antigens on cancer cells. Additionally, the heterogenous nature of most cancers makes single-antigen-targeting strategies suboptimal in many cases, leading to the development of innovative CAR T therapies designed to target multiple antigens. Furthermore, poor T cell trafficking and persistence contribute to the lack of success of CAR T cells against solid tumors. Addressing this challenge can be achieved through the local administration of T cells directly into the tumor site or the use of various strategies, such as using cytokines, to increase T cell persistence. Overall, cancer immunotherapy remains a promising field with significant potential to revolutionize cancer treatment, but further research is necessary to overcome the remaining challenges. In this review, we explored the mechanisms behind the challenges faced in the field of immunotherapy, including T cell exhaustion and the immunosuppressive nature of the TME. We also surveyed the therapeutic strategies utilized to combat cancer, with a focus on highlighting noteworthy and innovative emerging immunotherapies. We hope that this review will provide a useful resource for current and future professionals working in the field of cancer immunotherapy and related areas, enabling them to discover novel targets and create optimal and efficacious targeted treatments for cancer patients.
Author Contributions
C.C. and X.L. contributed to the conceptualization, research, writing, and review and editing of the manuscript. C.-Y.C. provided support in the research and drafting of the manuscript. H.Y.W. and R.-F.W. provided oversight and guidance throughout the process. All authors have read and agreed to the published version of the manuscript.
Funding
This work was in part supported by grants from the National Cancer Institute (NCI), National Institutes of Health (R01CA101795, R01CA246547, and U54CA210181), Department of Defense (DoD), Congressionally Directed Medical Research Programs (CDMRP) Breast Cancer Research Program (BCRP) (BC151081) and Lung Cancer Research Program (LCRP) (LC200368) to R-f.W.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.M.; Van Morris, K.; Vo, H.H.; Eck, S.; Lin, Y.F.; Rivas, J.M.; Andersson, B.S. T-cell receptor-based therapy: An innovative therapeutic approach for solid tumors. J. Hematol. Oncol. 2021, 14, 102. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Mullard, A. FDA approval of Immunocore’s first-in-class TCR therapeutic broadens depth of the T cell engager platform. Nat. Rev. Drug Discov. 2022, 21, 170. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, O.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [Green Version]
- Gorchakov, A.A.; Kulemzin, S.V.; Kochneva, G.V.; Taranin, A.V. Challenges and Prospects of Chimeric Antigen Receptor T-cell Therapy for Metastatic Prostate Cancer. Eur. Urol. 2020, 77, 299–308. [Google Scholar] [CrossRef]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurachi, M. CD8+ T cell exhaustion. Semin. Immunopathol. 2019, 41, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Thommen, D.S.; Schumacher, T.N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef] [Green Version]
- Straathof, K.; Flutter, B.; Wallace, R.; Jain, N.; Loka, T.; Depani, S.; Wright, G.; Thomas, S.; Cheung, G.W.; Gileadi, T.; et al. Antitumor activity without on-target off-tumor toxicity of GD2-chimeric antigen receptor T cells in patients with neuroblastoma. Sci. Transl. Med. 2020, 12, eabd6169. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.; Oeser, A.; Besiroglu, B.; Caro-Valenzuela, J.; Abd El Aziz, M.; Monsef, I.; Borchmann, P.; Estcourt, L.J.; Skoetz, N.; Goldkuhle, M. Chimeric antigen receptor (CAR) T-cell therapy for people with relapsed or refractory diffuse large B-cell lymphoma. Cochrane Database Syst. Rev. 2021, 9, CD013365. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Rosenberg, S.A. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat. Rev. Clin. Oncol. 2013, 10, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef] [Green Version]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef]
- Frey, N.V.; Shaw, P.A.; Hexner, E.O.; Pequignot, E.; Gill, S.; Luger, S.M.; Mangan, J.K.; Loren, A.W.; Perl, A.E.; Maude, S.L.; et al. Optimizing Chimeric Antigen Receptor T-Cell Therapy for Adults With Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2020, 38, 415–422. [Google Scholar] [CrossRef]
- Hassan, R.; Cohen, S.J.; Phillips, M.; Pastan, I.; Sharon, E.; Kelly, R.J.; Schweizer, C.; Weil, S.; Laheru, D. Phase I clinical trial of the chimeric anti-mesothelin monoclonal antibody MORAb-009 in patients with mesothelin-expressing cancers. Clin. Cancer Res. 2010, 16, 6132–6138. [Google Scholar] [CrossRef] [Green Version]
- Hassan, R.; Bullock, S.; Premkumar, A.; Kreitman, R.J.; Kindler, H.; Willingham, M.C.; Pastan, I. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin. Cancer Res. 2007, 13, 5144–5149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudley, M.E.; Wunderlich, J.; Nishimura, M.I.; Yu, D.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Hwu, P.; Marincola, F.M.; Sherry, R.; et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J. Immunother. 2001, 24, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.M.; Marincola, F.M.; Leitman, S.F.; Seipp, C.A.; et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J. Immunother. 2002, 25, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Van Bruggen, J.A.C.; Martens, A.W.J.; Fraietta, J.A.; Hofland, T.; Tonino, S.H.; Eldering, E.; Levin, M.D.; Siska, P.J.; Endstra, S.; Rathmell, J.C.; et al. Chronic lymphocytic leukemia cells impair mitochondrial fitness in CD8(+) T cells and impede CAR T-cell efficacy. Blood 2019, 134, 44–58. [Google Scholar] [CrossRef]
- Rosenberg, S.A. Raising the bar: The curative potential of human cancer immunotherapy. Sci. Transl. Med. 2012, 4, 127ps8. [Google Scholar] [CrossRef]
- Hinrichs, C.S.; Rosenberg, S.A. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol. Rev. 2014, 257, 56–71. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef]
- Gyurdieva, A.; Zajic, S.; Chang, Y.F.; Houseman, E.A.; Zhong, S.; Kim, J.; Nathenson, M.; Faitg, T.; Woessner, M.; Turner, D.C.; et al. Biomarker correlates with response to NY-ESO-1 TCR T cells in patients with synovial sarcoma. Nat. Commun. 2022, 13, 5296. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 (c259)T Cells in Synovial Sarcoma. Cancer Discov. 2018, 8, 944–957. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, I.; Lowther, D.E.; Dryer-Minnerly, R.; Wang, R.; Fayngerts, S.; Nunez, D.; Betts, G.; Bath, N.; Tipping, A.J.; Melchiori, L.; et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J. Immunother. Cancer 2019, 7, 276. [Google Scholar] [CrossRef] [PubMed]
- Abate-Daga, D.; Hanada, K.; Davis, J.L.; Yang, J.C.; Rosenberg, S.A.; Morgan, R.A. Expression profiling of TCR-engineered T cells demonstrates overexpression of multiple inhibitory receptors in persisting lymphocytes. Blood 2013, 122, 1399–1410. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, Y.; Eliyahu, S.; Delgado, C.H.; Robbins, P.F.; Rivoltini, L.; Topalian, S.L.; Miki, T.; Rosenberg, S.A. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc. Natl. Acad. Sci. USA 1994, 91, 3515–3519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, Y.; Eliyahu, S.; Delgado, C.H.; Robbins, P.F.; Sakaguchi, K.; Appella, E.; Yannelli, J.R.; Adema, G.J.; Miki, T.; Rosenberg, S.A. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc. Natl. Acad. Sci. USA 1994, 91, 6458–6462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, Y.; Eliyahu, S.; Sakaguchi, K.; Robbins, P.F.; Rivoltini, L.; Yannelli, J.R.; Appella, E.; Rosenberg, S.A. Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2-restricted tumor infiltrating lymphocytes. J. Exp. Med. 1994, 180, 347–352. [Google Scholar] [CrossRef] [Green Version]
- Allison, J.P.; McIntyre, B.W.; Bloch, D. Tumor-specific antigen of murine T-lymphoma defined with monoclonal antibody. J. Immunol. 1982, 129, 2293–2300. [Google Scholar] [CrossRef] [PubMed]
- Fabbi, M.; Acuto, O.; Smart, J.E.; Reinherz, E.L. Homology of Ti alpha-subunit of a T-cell antigen-MHC receptor with immunoglobulin. Nature 1984, 312, 269–271. [Google Scholar] [CrossRef]
- Hedrick, S.M.; Cohen, D.I.; Nielsen, E.A.; Davis, M.M. Isolation of cDNA clones encoding T cell-specific membrane-associated proteins. Nature 1984, 308, 149–153. [Google Scholar] [CrossRef]
- Morath, A.; Schamel, W.W. alphabeta and gammadelta T cell receptors: Similar but different. J. Leukoc. Biol. 2020, 107, 1045–1055. [Google Scholar] [CrossRef]
- Call, M.E.; Pyrdol, J.; Wiedmann, M.; Wucherpfennig, K.W. The organizing principle in the formation of the T cell receptor-CD3 complex. Cell 2002, 111, 967–979. [Google Scholar] [CrossRef] [Green Version]
- Dushek, O.; Goyette, J.; van der Merwe, P.A. Non-catalytic tyrosine-phosphorylated receptors. Immunol. Rev. 2012, 250, 258–276. [Google Scholar] [CrossRef] [PubMed]
- Huse, M. The T-cell-receptor signaling network. J. Cell Sci. 2009, 122, 1269–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Kadlecek, T.A.; Au-Yeung, B.B.; Goodfellow, H.E.; Hsu, L.Y.; Freedman, T.S.; Weiss, A. ZAP-70: An essential kinase in T-cell signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, A.C.; Irving, B.A.; Fraser, J.D.; Weiss, A. The zeta chain is associated with a tyrosine kinase and upon T-cell antigen receptor stimulation associates with ZAP-70, a 70-kDa tyrosine phosphoprotein. Proc. Natl. Acad. Sci. USA 1991, 88, 9166–9170. [Google Scholar] [CrossRef] [Green Version]
- Balagopalan, L.; Coussens, N.P.; Sherman, E.; Samelson, L.E.; Sommers, C.L. The LAT story: A tale of cooperativity, coordination, and choreography. Cold Spring Harb. Perspect. Biol. 2010, 2, a005512. [Google Scholar] [CrossRef] [Green Version]
- Bunnell, S.C.; Diehn, M.; Yaffe, M.B.; Findell, P.R.; Cantley, L.C.; Berg, L.J. Biochemical interactions integrating Itk with the T cell receptor-initiated signaling cascade. J. Biol. Chem. 2000, 275, 2219–2230. [Google Scholar] [CrossRef] [Green Version]
- Martinez, G.J.; Pereira, R.M.; Aijo, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Shayan, G.; Srivastava, R.; Li, J.; Schmitt, N.; Kane, L.P.; Ferris, R.L. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. OncoImmunology 2017, 6, e1261779. [Google Scholar] [CrossRef] [Green Version]
- Jeon, H.-S.; Jin, G.; Kang, H.-G.; Choi, Y.Y.; Lee, W.K.; Choi, J.E.; Bae, E.Y.; Yoo, S.S.; Lee, S.Y.; Lee, E.B.; et al. A functional variant at 19q13.3, rs967591G>A, is associated with shorter survival of early-stage lung cancer. Clin. Cancer Res. 2013, 19, 4185–4195. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Choi, J.E.; Jeon, H.-S.; Choi, Y.-Y.; Lee, W.K.; Lee, E.B.; Lee, H.C.; Kang, H.-G.; Yoo, S.S.; Lee, J.; et al. A Panel of Genetic Polymorphism for the Prediction of Prognosis in Patients with Early Stage Non-Small Cell Lung Cancer after Surgical Resection. PLoS ONE 2015, 10, e0140216. [Google Scholar] [CrossRef] [Green Version]
- Thommen, D.S.; Schreiner, J.; Müller, P.; Herzig, P.; Roller, A.; Belousov, A.; Umana, P.; Pisa, P.; Klein, C.; Bacac, M.; et al. Progression of Lung Cancer Is Associated with Increased Dysfunction of T Cells Defined by Coexpression of Multiple Inhibitory Receptors. Cancer Immunol. Res. 2015, 3, 1344–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Zhang, Y.; Zheng, L.; Zheng, C.; Song, J.; Zhang, Q.; Kang, B.; Liu, Z.; Jin, L.; Xing, R.; et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018, 24, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Xu, J.; Ni, J.; Gao, X.; Zhu, Z.; Dong, D.; Wang, X.; Shi, C.; Tao, X.; Dong, W.; et al. A functional insertion/deletion polymorphism in the proximal promoter of CD3G is associated with susceptibility for hepatocellular carcinoma in Chinese population. DNA Cell Biol. 2012, 31, 1480–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, C.; Zheng, L.; Yoo, J.-K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; He, Q.; Shen, H.; Xia, A.; Tian, W.; Yu, W.; Sun, B. TOX promotes the exhaustion of antitumor CD8+ T cells by preventing PD1 degradation in hepatocellular carcinoma. J. Hepatol. 2019, 71, 731–741. [Google Scholar] [CrossRef]
- Peña-Romero, A.C.; Orenes-Piñero, E. Dual Effect of Immune Cells within Tumour Microenvironment: Pro- and Anti-Tumour Effects and Their Triggers. Cancers 2022, 14, 1681. [Google Scholar] [CrossRef]
- Liang, C.; Huang, S.; Zhao, Y.; Chen, S.; Li, Y. TOX as a potential target for immunotherapy in lymphocytic malignancies. Biomarker Res. 2021, 9, 20. [Google Scholar] [CrossRef]
- Lu, X.; Yang, L.; Yao, D.; Wu, X.; Li, J.; Liu, X.; Deng, L.; Huang, C.; Wang, Y.; Li, D.; et al. Tumor antigen-specific CD8(+) T cells are negatively regulated by PD-1 and Tim-3 in human gastric cancer. Cell Immunol. 2017, 313, 43–51. [Google Scholar] [CrossRef]
- Tycko, B.; Smith, S.D.; Sklar, J. Chromosomal translocations joining LCK and TCRB loci in human T cell leukemia. J. Exp. Med. 1991, 174, 867–873. [Google Scholar] [CrossRef] [Green Version]
- Rassenti, L.Z.; Huynh, L.; Toy, T.L.; Chen, L.; Keating, M.J.; Gribben, J.G.; Neuberg, D.S.; Flinn, I.W.; Rai, K.R.; Byrd, J.C.; et al. ZAP-70 compared with immunoglobulin heavy-chain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. N. Engl. J. Med. 2004, 351, 893–901. [Google Scholar] [CrossRef]
- Ruiz, I.; Vicario, R.; Morancho, B.; Morales, C.; Arenas, E.; Herter, S.; Freimoser-Grundschober, A.; Somandin, J.; Sam, J.; Ast, O.; et al. p95HER2–T cell bispecific antibody for breast. Sci. Transl. Med. 2018, 10, eaat1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zippelius, A.; Batard, P.; Rubio-Godoy, V.; Bioley, G.; Liénard, D.; Lejeune, F.; Rimoldi, D.; Guillaume, P.; Meidenbauer, N.; Mackensen, A.; et al. Effector Function of Human Tumor-Specific CD8 T Cells in Melanoma Lesions: A State of Local Functional Tolerance. Cancer Res. 2004, 64, 2865–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Baitsch, L.; Baumgaertner, P.; Devêvre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of tumor-specific CD8⁺ T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallois, D.; Dobay, M.P.D.; Morin, R.D.; Lemonnier, F.; Missiaglia, E.; Juilland, M.; Iwaszkiewicz, J.; Fataccioli, V.; Bisig, B.; Roberti, A.; et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood 2016, 128, 1490–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaqué, J.P.; Gómez-López, G.; Monsálvez, V.; Varela, I.; Martínez, N.; Pérez, C.; Domínguez, O.; Graña, O.; Rodríguez-Peralto, J.L.; Rodríguez-Pinilla, S.M.; et al. PLCG1 mutations in cutaneous T-cell lymphomas. Blood 2014, 123, 2034–2043. [Google Scholar] [CrossRef]
- Hayes, T.K.; Neel, N.F.; Hu, C.; Gautam, P.; Chenard, M.; Long, B.; Aziz, M.; Kassner, M.; Bryant, K.L.; Pierobon, M.; et al. Long-Term ERK Inhibition in KRAS-Mutant Pancreatic Cancer Is Associated with MYC Degradation and Senescence-like Growth Suppression. Cancer Cell 2016, 29, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, G.A.; Wittinghofer, A.; Der, C.J. Selective Targeting of the KRAS G12C Mutant: Kicking KRAS When It’s Down. Cancer Cell 2016, 29, 251–253. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.Y.; Brown, L.; Stevenson, K.; deSouza, T.; Aster, J.C.; Louissaint, A., Jr.; Weinstock, D.M. RhoA G17V is sufficient to induce autoimmunity and promotes T-cell lymphomagenesis in mice. Blood 2018, 132, 935–947. [Google Scholar] [CrossRef] [Green Version]
- Barreira, M.; Fabbiano, S.; Couceiro, J.R.; Torreira, E.; Martínez-Torrecuadrada, J.L.; Montoya, G.; Llorca, O.; Bustelo, X.R. The C-terminal SH3 domain contributes to the intramolecular inhibition of Vav family proteins. Sci. Signal. 2014, 7, ra35. [Google Scholar] [CrossRef] [Green Version]
- Shah, K.; Al-Haidari, A.; Sun, J.; Kazi, J.U. T cell receptor (TCR) signaling in health and disease. Sig. Transduct. Target Ther. 2021, 6, 412. [Google Scholar] [CrossRef] [PubMed]
- Linka, R.M.; Risse, S.L.; Bienemann, K.; Werner, M.; Linka, Y.; Krux, F.; Synaeve, C.; Deenen, R.; Ginzel, S.; Dvorsky, R.; et al. Loss-of-function mutations within the IL-2 inducible kinase ITK in patients with EBV-associated lymphoproliferative diseases. Leukemia 2012, 26, 963–971. [Google Scholar] [CrossRef]
- Robles-Valero, J.; Lorenzo-Martín, L.F.; Menacho-Márquez, M.; Fernández-Pisonero, I.; Abad, A.; Camós, M.; Toribio, M.L.; Espinosa, L.; Bigas, A.; Bustelo, X.R. A Paradoxical Tumor-Suppressor Role for the Rac1 Exchange Factor Vav1 in T Cell Acute Lymphoblastic Leukemia. Cancer Cell 2017, 32, 608–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohr, J.; Guo, S.; Huo, J.; Bouska, A.; Lachel, C.; Li, Y.; Simone, P.D.; Zhang, W.; Gong, Q.; Wang, C.; et al. Recurrent activating mutations of CD28 in peripheral T-cell lymphomas. Leukemia 2016, 30, 1062–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quivoron, C.; Couronne, L.; Della Valle, V.; Lopez, C.K.; Plo, I.; Wagner-Ballon, O.; Do Cruzeiro, M.; Delhommeau, F.; Arnulf, B.; Stern, M.H.; et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 2011, 20, 25–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, R.A.; Iqbal, J.; Lemonnier, F.; Kucuk, C.; de Leval, L.; Jais, J.-P.; Parrens, M.; Martin, A.; Xerri, L.; Brousset, P.; et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood 2012, 119, 1901–1903. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; McKeithan, T.W.; Gong, Q.; Zhang, W.; Bouska, A.; Rosenwald, A.; Gascoyne, R.D.; Wu, X.; Wang, J.; Muhammad, Z.; et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood 2015, 126, 1741–1752. [Google Scholar] [CrossRef] [Green Version]
- Palomero, T.; Couronné, L.; Khiabanian, H.; Kim, M.-Y.; Ambesi-Impiombato, A.; Perez-Garcia, A.; Carpenter, Z.; Abate, F.; Allegretta, M.; Haydu, J.E.; et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat. Genet. 2014, 46, 166–170. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, J.S.; Kim, J.; Kim, S.J.; Kim, W.S.; Lee, S.; Ko, Y.H.; Yoo, H.Y. A highly recurrent novel missense mutation in CD28 among angioimmunoblastic T-cell lymphoma patients. Haematologica 2015, 100, e505–e507. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, H.; O’Shea, J.J.; Longo, D.L.; Loeffler, C.M.; McVicar, D.W.; Ochoa, A.C. Alterations in signal transduction molecules in T lymphocytes from tumor-bearing mice. Science 1992, 258, 1795–1798. [Google Scholar] [CrossRef]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, E.; Zhang, Y.; Zhu, G.; Hao, J.; Persson, X.-M.T.; Egilmez, N.K.; Suttles, J.; Li, B. Deficiency of AMPK in CD8+ T cells suppresses their anti-tumor function by inducing protein phosphatase-mediated cell death. Oncotarget 2019, 6, 7944–7958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Perica, K.; Klebanoff, C.A.; Wolchok, J.D. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2022, 19, 775–790. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zhang, Y.; Hu, Y.; Mei, H. T Cell Exhaustion and CAR-T Immunotherapy in Hematological Malignancies. Biomed. Res. Int. 2021, 2021, 6616391. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wu, K.; Tao, K.; Chen, L.; Zheng, Q.; Lu, X.; Liu, J.; Shi, L.; Liu, C.; Wang, G.; et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology 2012, 56, 1342–1351. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Kuchroo, V.K.; Anderson, A.C.; Petrovas, C. Coinhibitory receptors and CD8 T cell exhaustion in chronic infections. Curr. Opin. HIV AIDS 2014, 9, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Leun, A.M.; Thommen, D.S.; Schumacher, T.N. CD8+ T cell states in human cancer: Insights from single-cell analysis. Nat. Rev. Cancer 2020, 20, 218–232. [Google Scholar] [CrossRef]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Farber, D.L. Form and function for T cells in health and disease. Nat. Rev. Immunol. 2020, 20, 83–84. [Google Scholar] [CrossRef]
- Schnell, A.; Bod, L.; Madi, A.; Kuchroo, V.K. The yin and yang of co-inhibitory receptors: Toward anti-tumor immunity without autoimmunity. Cell Res. 2020, 30, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Zebley, C.C.; Brown, C.; Mi, T.; Fan, Y.; Alli, S.; Boi, S.; Galletti, G.; Lugli, E.; Langfitt, D.; Metais, J.Y.; et al. CD19-CAR T cells undergo exhaustion DNA methylation programming in patients with acute lymphoblastic leukemia. Cell Rep. 2021, 37, 110079. [Google Scholar] [CrossRef]
- Kansy, B.A.; Concha-Benavente, F.; Srivastava, R.M.; Jie, H.B.; Shayan, G.; Lei, Y.; Moskovitz, J.; Moy, J.; Li, J.; Brandau, S.; et al. PD-1 Status in CD8(+) T Cells Associates with Survival and Anti-PD-1 Therapeutic Outcomes in Head and Neck Cancer. Cancer Res. 2017, 77, 6353–6364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, W.; Dimitri, A.; Wang, W.; Jung, I.Y.; Ott, C.J.; Fasolino, M.; Wang, Y.; Kulikovskaya, I.; Gupta, M.; Yoder, T.; et al. BET bromodomain protein inhibition reverses chimeric antigen receptor extinction and reinvigorates exhausted T cells in chronic lymphocytic leukemia. J. Clin. Investig. 2021, 131, e145459. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Han, G.; Puebla-Osorio, N.; Ma, M.C.J.; Strati, P.; Chasen, B.; Dai, E.; Dang, M.; Jain, N.; Yang, H.; et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat. Med. 2020, 26, 1878–1887. [Google Scholar] [CrossRef] [PubMed]
- Vahteristo, P.; Bartkova, J.; Eerola, H.; Syrjakoski, K.; Ojala, S.; Kilpivaara, O.; Tamminen, A.; Kononen, J.; Aittomaki, K.; Heikkila, P.; et al. A CHEK2 genetic variant contributing to a substantial fraction of familial breast cancer. Am. J. Hum. Genet. 2002, 71, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Guan, Q.; Wang, Y.; Zhao, Z.J.; Zhou, G.W. SHP-1 suppresses cancer cell growth by promoting degradation of JAK kinases. J. Cell Biochem. 2003, 90, 1026–1037. [Google Scholar] [CrossRef]
- Wu, C.; Sun, M.; Liu, L.; Zhou, G.W. The function of the protein tyrosine phosphatase SHP-1 in cancer. Gene 2003, 306, 1–12. [Google Scholar] [CrossRef]
- Zhang, T.; Guo, W.; Yang, Y.; Liu, W.; Guo, L.; Gu, Y.; Shu, Y.; Wang, L.; Wu, X.; Hua, Z.; et al. Loss of SHP-2 activity in CD4+ T cells promotes melanoma progression and metastasis. Sci. Rep. 2013, 3, 2845. [Google Scholar] [CrossRef] [PubMed]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.L.; Camara, S.; Shakiba, M.; Scott, A.C.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef] [Green Version]
- Mognol, G.P.; Spreafico, R.; Wong, V.; Scott-Browne, J.P.; Togher, S.; Hoffmann, A.; Hogan, P.G.; Rao, A.; Trifari, S. Exhaustion-associated regulatory regions in CD8+ tumor-infiltrating T cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2776–E2785. [Google Scholar] [CrossRef] [Green Version]
- Soto-Nieves, N.; Puga, I.; Abe, B.T.; Bandyopadhyay, S.; Baine, I.; Rao, A.; Macian, F. Transcriptional complexes formed by NFAT dimers regulate the induction of T cell tolerance. J. Exp. Med. 2009, 206, 867–876. [Google Scholar] [CrossRef] [Green Version]
- Harant, H.; Lindley, I.J.D. Negative cross-talk between the human orphan nuclear receptor Nur77/NAK-1/TR3 and nuclear factor-κB. Nucleic Acids Res. 2004, 32, 5280–5290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wang, Y.; Lu, H.; Li, J.; Yan, X.; Xiao, M.; Hao, J.; Alekseev, A.; Khong, H.; Chen, T.; et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 2019, 567, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Cheng, S.; Luo, N.; Gao, R.; Yu, K.; Kang, B.; Wang, L.; Zhang, Q.; Fang, Q.; Zhang, L.; et al. Distinct epigenetic features of tumor-reactive CD8+ T cells in colorectal cancer patients revealed by genome-wide DNA methylation analysis. Genome Biol. 2019, 21, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; López-Moyado, I.F.; Seo, H.; Lio, C.-W.J.; Hempleman, L.J.; Sekiya, T.; Yoshimura, A.; Scott-Browne, J.P.; Rao, A. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 2019, 567, 530–534. [Google Scholar] [CrossRef]
- Odagiu, L.; May, J.; Boulet, S.; Baldwin, T.A.; Labrecque, N. Role of the Orphan Nuclear Receptor NR4A Family in T-Cell Biology. Front. Endocrinol. 2020, 11, 624122. [Google Scholar] [CrossRef]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [Green Version]
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef]
- Bordon, Y. TOX for tired T cells. Nat. Rev. Immunol. 2019, 19, 476. [Google Scholar] [CrossRef]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Chen, J.; Gonzalez-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; Lopez-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef] [Green Version]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gacerez, A.T.; Sentman, C.L. T-bet promotes potent antitumor activity of CD4(+) CAR T cells. Cancer Gene Ther. 2018, 25, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.H.; Pages, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haabeth, O.A.; Lorvik, K.B.; Hammarstrom, C.; Donaldson, I.M.; Haraldsen, G.; Bogen, B.; Corthay, A. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat. Commun. 2011, 2, 240. [Google Scholar] [CrossRef] [Green Version]
- Kallies, A.; Good-Jacobson, K.L. Transcription Factor T-bet Orchestrates Lineage Development and Function in the Immune System. Trends Immunol. 2017, 38, 287–297. [Google Scholar] [CrossRef]
- Li, J.; Shayan, G.; Avery, L.; Jie, H.-B.; Gildener-Leapman, N.; Schmitt, N.; Lu, B.F.; Kane, L.P.; Ferris, R.L. Tumor-infiltrating Tim-3+ T cells proliferate avidly except when PD-1 is co-expressed: Evidence for intracellular cross talk. OncoImmunology 2016, 5, e1200778. [Google Scholar] [CrossRef] [Green Version]
- Kao, C.; Oestreich, K.J.; Paley, M.A.; Crawford, A.; Angelosanto, J.M.; Ali, M.A.; Intlekofer, A.M.; Boss, J.M.; Reiner, S.L.; Weinmann, A.S.; et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat. Immunol. 2011, 12, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Ha, S.-J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; He, Y.; Hao, J.; Ni, L.; Dong, C. High Levels of Eomes Promote Exhaustion of Anti-tumor CD8(+) T Cells. Front. Immunol. 2018, 9, 2981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, K.; Gabriel, S.S.; Liao, Y.; Gloury, R.; Preston, S.; Henstridge, D.C.; Pellegrini, M.; Zehn, D.; Berberich-Siebelt, F.; Febbraio, M.A.; et al. Transcription Factor IRF4 Promotes CD8(+) T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 2017, 47, 1129–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, H.; Gonzalez-Avalos, E.; Zhang, W.; Ramchandani, P.; Yang, C.; Lio, C.J.; Rao, A.; Hogan, P.G. BATF and IRF4 cooperate to counter exhaustion in tumor-infiltrating CAR T cells. Nat. Immunol. 2021, 22, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Yue, T.-T.; Yang, C.-L.; Wang, F.-X.; Luo, J.-H.; Rong, S.-J.; Zhang, M.; Guo, Y.; Xiong, F.; Wang, C.-Y. The MAPK dual specific phosphatase (DUSP) proteins: A versatile wrestler in T cell functionality. Int. Immunopharmacol. 2021, 98, 107906. [Google Scholar] [CrossRef]
- Lu, D.; Liu, L.; Sun, Y.Z.; Song, J.; Yin, Q.; Zhang, G.Z.; Qi, F.; Hu, Z.X.; Yang, Z.L.; Zhou, Z.; et al. The phosphatase PAC1 acts as a T cell suppressor and attenuates host antitumor immunity. Nat. Immunol. 2020, 21, 287–297. [Google Scholar] [CrossRef]
- Lissbrant, I.F.; Stattin, P.; Wikstrom, P.; Damber, J.E.; Egevad, L.; Bergh, A. Tumor associated macrophages in human prostate cancer: Relation to clinicopathological variables and survival. Int. J. Oncol. 2000, 17, 445–451. [Google Scholar] [CrossRef]
- Wiede, F.; Lu, K.H.; Du, X.; Liang, S.; Hochheiser, K.; Dodd, G.T.; Goh, P.K.; Kearney, C.; Meyran, D.; Beavis, P.A.; et al. PTPN2 phosphatase deletion in T cells promotes anti-tumour immunity and CAR T-cell efficacy in solid tumours. EMBO J. 2020, 39, e103637. [Google Scholar] [CrossRef]
- Ten Hoeve, J.; de Jesus Ibarra-Sanchez, M.; Fu, Y.; Zhu, W.; Tremblay, M.; David, M.; Shuai, K. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol. Cell. Biol. 2002, 22, 5662–5668. [Google Scholar] [CrossRef] [Green Version]
- Simoncic, P.D.; Lee-Loy, A.; Barber, D.L.; Tremblay, M.L.; McGlade, C.J. The T cell protein tyrosine phosphatase is a negative regulator of janus family kinases 1 and 3. Curr. Biol. 2002, 12, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Wiede, F.; Dudakov, J.A.; Lu, K.H.; Dodd, G.T.; Butt, T.; Godfrey, D.I.; Strasser, A.; Boyd, R.L.; Tiganis, T. PTPN2 regulates T cell lineage commitment and alphabeta versus gammadelta specification. J. Exp. Med. 2017, 214, 2733–2758. [Google Scholar] [CrossRef]
- LaFleur, M.W.; Nguyen, T.H.; Coxe, M.A.; Miller, B.C.; Yates, K.B.; Gillis, J.E.; Sen, D.R.; Gaudiano, E.F.; Al Abosy, R.; Freeman, G.J.; et al. PTPN2 regulates the generation of exhausted CD8(+) T cell subpopulations and restrains tumor immunity. Nat. Immunol. 2019, 20, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, C.; Bukczynska, P.E.; Puryer, M.A.; Sadek, C.M.; Shields, B.J.; Tremblay, M.L.; Tiganis, T. Selective regulation of tumor necrosis factor-induced Erk signaling by Src family kinases and the T cell protein tyrosine phosphatase. Nat. Immunol. 2005, 6, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Odejide, O.; Weigert, O.; Lane, A.A.; Toscano, D.; Lunning, M.A.; Kopp, N.; Kim, S.; van Bodegom, D.; Bolla, S.; Schatz, J.H.; et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood 2014, 123, 1293–1296. [Google Scholar] [CrossRef]
- Feng, Y.; Li, X.; Cassady, K.; Zou, Z.; Zhang, X. TET2 Function in Hematopoietic Malignancies, Immune Regulation, and DNA Repair. Front. Oncol. 2019, 9, 210. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; van der Leun, A.; Yofe, I.; Lubling, Y.; Solodkin, D.G.; van Akkooi, A.; van den Braber, M.; Rozeman, L.; Haanen, J.; Blank, C.; et al. Dysfunctional CD8+ T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 2019, 176, 775–789. [Google Scholar] [CrossRef]
- Shin, H.; Blackburn, S.D.; Intlekofer, A.M.; Kao, C.; Angelosanto, J.M.; Reiner, S.L.; Wherry, E.J. A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity 2009, 31, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Frenzel, A.; Grespi, F.; Chmelewskij, W.; Villunger, A. Bcl2 family proteins in carcinogenesis and the treatment of cancer. Apoptosis 2009, 14, 584–596. [Google Scholar] [CrossRef] [Green Version]
- Ren, R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer 2005, 5, 172–183. [Google Scholar] [CrossRef]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Vliagoftis, H.; Worobec, A.S.; Metcalfe, D.D. The protooncogene c-kit and c-kit ligand in human disease. J. Allergy Clin. Immunol. 1997, 100, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, R.I.; Gee, J.M.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37 (Suppl. 4), S9–S15. [Google Scholar] [CrossRef] [PubMed]
- Doherty, L.; Gigas, D.C.; Kesari, S.; Drappatz, J.; Kim, R.; Zimmerman, J.; Ostrowsky, L.; Wen, P.Y. Pilot study of the combination of EGFR and mTOR inhibitors in recurrent malignant gliomas. Neurology 2006, 67, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.M.; Dominguez, G.; Gonzalez-Sancho, J.M.; Garcia, J.M.; Silva, J.; Garcia-Andrade, C.; Navarro, A.; Munoz, A.; Bonilla, F. Expression of thyroid hormone receptor/erbA genes is altered in human breast cancer. Oncogene 2002, 21, 4307–4316. [Google Scholar] [CrossRef] [Green Version]
- Amano, R.; Namekata, M.; Horiuchi, M.; Saso, M.; Yanagisawa, T.; Tanaka, Y.; Ghani, F.I.; Yamamoto, M.; Sakamoto, T. Specific inhibition of FGF5-induced cell proliferation by RNA aptamers. Sci. Rep. 2021, 11, 2976. [Google Scholar] [CrossRef]
- Hanada, K.; Yewdell, J.W.; Yang, J.C. Immune recognition of a human renal cancer antigen through post-translational protein splicing. Nature 2004, 427, 252–256. [Google Scholar] [CrossRef]
- El-Gamal, M.I.; Anbar, H.S.; Yoo, K.H.; Oh, C.H. FMS Kinase Inhibitors: Current Status and Future Prospects. Med. Res. Rev. 2013, 33, 599–636. [Google Scholar] [CrossRef]
- Goustin, A.S.; Leof, E.B.; Shipley, G.D.; Moses, H.L. Growth factors and cancer. Cancer Res. 1986, 46, 1015–1029. [Google Scholar]
- Ruiz i Altaba, A. Gli proteins and Hedgehog signaling: Development and cancer. Trends Genet. 1999, 15, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y. Biology of HER2 and its importance in breast cancer. Oncology 2001, 61 (Suppl. 2), 1–13. [Google Scholar] [CrossRef]
- Nishihira, T.; Hashimoto, Y.; Katayama, M.; Mori, S.; Kuroki, T. Molecular and cellular features of esophageal cancer cells. J. Cancer Res. Clin. Oncol. 1993, 119, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Nateri, A.S.; Spencer-Dene, B.; Behrens, A. Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature 2005, 437, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Natali, P.G.; Prat, M.; Nicotra, M.R.; Bigotti, A.; Olivero, M.; Comoglio, P.M.; Di Renzo, M.F. Overexpression of the met/HGF receptor in renal cell carcinomas. Int. J. Cancer 1996, 69, 212–217. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Balmain, A.; Counter, C.M. A model for RAS mutation patterns in cancers: Finding the sweet spot. Nat. Rev. Cancer 2018, 18, 767–777. [Google Scholar] [CrossRef]
- Yunis, J.J.; Boot, A.J.; Mayer, M.G.; Bos, J.L. Mechanisms of ras mutation in myelodysplastic syndrome. Oncogene 1989, 4, 609–614. [Google Scholar]
- Riely, G.J.; Marks, J.; Pao, W. KRAS mutations in non-small cell lung cancer. Proc. Am. Thorac. Soc. 2009, 6, 201–205. [Google Scholar] [CrossRef]
- Locker, G.Y.; Hamilton, S.; Harris, J.; Jessup, J.M.; Kemeny, N.; Macdonald, J.S.; Somerfield, M.R.; Hayes, D.F.; Bast, R.C., Jr.; Asco. ASCO 2006 update of recommendations for the use of tumor markers in gastrointestinal cancer. J. Clin. Oncol. 2006, 24, 5313–5327. [Google Scholar] [CrossRef] [PubMed]
- Arighi, E.; Borrello, M.G.; Sariola, H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev. 2005, 16, 441–467. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, A.; Heldin, C.H.; Wasteson, A.; Westermark, B.; Deuel, T.F.; Huang, J.S.; Seeburg, P.H.; Gray, A.; Ullrich, A.; Scrace, G.; et al. The c-sis gene encodes a precursor of the B chain of platelet-derived growth factor. EMBO J. 1984, 3, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Kharbanda, S.; Kufe, D. Association of the DF3/MUC1 breast cancer antigen with Grb2 and the Sos/Ras exchange protein. Cancer Res. 1995, 55, 4000–4003. [Google Scholar] [PubMed]
- Wheeler, D.L.; Iida, M.; Dunn, E.F. The role of Src in solid tumors. Oncologist 2009, 14, 667–678. [Google Scholar] [CrossRef]
- Galway, M.E.; Masucci, J.D.; Lloyd, A.M.; Walbot, V.; Davis, R.W.; Schiefelbein, J.W. The TTG gene is required to specify epidermal cell fate and cell patterning in the Arabidopsis root. Dev. Biol. 1994, 166, 740–754. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- LeCouter, J.; Moritz, D.R.; Li, B.; Phillips, G.L.; Liang, X.H.; Gerber, H.P.; Hillan, K.J.; Ferrara, N. Angiogenesis-independent endothelial protection of liver: Role of VEGFR-1. Science 2003, 299, 890–893. [Google Scholar] [CrossRef]
- Sitohy, B.; Nagy, J.A.; Dvorak, H.F. Anti-VEGF/VEGFR therapy for cancer: Reassessing the target. Cancer Res. 2012, 72, 1909–1914. [Google Scholar] [CrossRef] [Green Version]
- Cooper, G.M. The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates Inc.: Sunderland, MA, USA, 2000; p. 689. [Google Scholar]
- Wang, L.H.; Wu, C.F.; Rajasekaran, N.; Shin, Y.K. Loss of Tumor Suppressor Gene Function in Human Cancer: An Overview. Cell. Physiol. Biochem. 2018, 51, 2647–2693. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef]
- Jayaprakash, P.; Ai, M.; Liu, A.; Budhani, P.; Bartkowiak, T.; Sheng, J.; Ager, C.; Nicholas, C.; Jaiswal, A.R.; Sun, Y.; et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Investig. 2018, 128, 5137–5149. [Google Scholar] [CrossRef] [PubMed]
- Gerald, W.L.; Gramling, T.S.; Sens, D.A.; Garvin, A.J. Expression of the 11p13 Wilms’ tumor gene, WT1, correlates with histologic category of Wilms’ tumor. Am. J. Pathol. 1992, 140, 1031–1037. [Google Scholar]
- De Carvalho, D.D.; You, J.S.; Jones, P.A. DNA methylation and cellular reprogramming. Trends Cell Biol. 2010, 20, 609–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanai, Y.; Ushijima, S.; Nakanishi, Y.; Sakamoto, M.; Hirohashi, S. Mutation of the DNA methyltransferase (DNMT) 1 gene in human colorectal cancers. Cancer Lett. 2003, 192, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Strawn, E.; Basir, Z.; Halverson, G.; Guo, S.W. Aberrant expression of deoxyribonucleic acid methyltransferases DNMT1, DNMT3A, and DNMT3B in women with endometriosis. Fertil. Steril. 2007, 87, 24–32. [Google Scholar] [CrossRef]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, Y.; Yuan, J.; Suetake, I.; Suzuki, H.; Ishikawa, Y.; Choi, Y.L.; Ueno, T.; Soda, M.; Hamada, T.; Haruta, H.; et al. Array-based genomic resequencing of human leukemia. Oncogene 2010, 29, 3723–3731. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.J.; Xu, J.; Gu, Z.H.; Pan, C.M.; Lu, G.; Shen, Y.; Shi, J.Y.; Zhu, Y.M.; Tang, L.; Zhang, X.W.; et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet. 2011, 43, 309–315. [Google Scholar] [CrossRef]
- Wijmenga, C.; Hansen, R.S.; Gimelli, G.; Bjorck, E.J.; Davies, E.G.; Valentine, D.; Belohradsky, B.H.; van Dongen, J.J.; Smeets, D.F.; van den Heuvel, L.P.; et al. Genetic variation in ICF syndrome: Evidence for genetic heterogeneity. Hum. Mutat. 2000, 16, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.M.; Ohno, K.; Fukudome, T.; Tsujino, A.; Brengman, J.M.; De Vivo, D.C.; Packer, R.J.; Engel, A.G. Congenital myasthenic syndrome caused by low-expressor fast-channel AChR delta subunit mutation. Neurology 2002, 59, 1881–1888. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansom, O.J.; Maddison, K.; Clarke, A.R. Mechanisms of disease: Methyl-binding domain proteins as potential therapeutic targets in cancer. Nat. Clin. Pract. Oncol. 2007, 4, 305–315. [Google Scholar] [CrossRef]
- Gui, Y.; Guo, G.; Huang, Y.; Hu, X.; Tang, A.; Gao, S.; Wu, R.; Chen, C.; Li, X.; Zhou, L.; et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 2011, 43, 875–878. [Google Scholar] [CrossRef]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Li, J.; Song, L. Bmi-1, stem cells and cancer. Acta Biochim. Biophys. Sin. 2009, 41, 527–534. [Google Scholar] [CrossRef] [Green Version]
- Lukacs, R.U.; Memarzadeh, S.; Wu, H.; Witte, O.N. Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell Stem Cell 2010, 7, 682–693. [Google Scholar] [CrossRef] [Green Version]
- Miremadi, A.; Oestergaard, M.Z.; Pharoah, P.D.; Caldas, C. Cancer genetics of epigenetic genes. Hum. Mol. Genet. 2007, 16, R28–R49. [Google Scholar] [CrossRef] [PubMed]
- Chase, A.; Cross, N.C. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011, 17, 2613–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, D.P.; Cheng, A.S. Epigenetic regulation of signaling pathways in cancer: Role of the histone methyltransferase EZH2. J. Gastroenterol. Hepatol. 2011, 26, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Varier, R.A.; Timmers, H.T. Histone lysine methylation and demethylation pathways in cancer. Biochim. Biophys. Acta 2011, 1815, 75–89. [Google Scholar] [CrossRef]
- Ropero, S.; Fraga, M.F.; Ballestar, E.; Hamelin, R.; Yamamoto, H.; Boix-Chornet, M.; Caballero, R.; Alaminos, M.; Setien, F.; Paz, M.F.; et al. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat. Genet. 2006, 38, 566–569. [Google Scholar] [CrossRef]
- Rotili, D.; Mai, A. Targeting Histone Demethylases: A New Avenue for the Fight against Cancer. Genes Cancer 2011, 2, 663–679. [Google Scholar] [CrossRef]
- Jones, S.; Wang, T.L.; Shih Ie, M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A., Jr.; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Guan, B.; Mao, T.L.; Panuganti, P.K.; Kuhn, E.; Kurman, R.J.; Maeda, D.; Chen, E.; Jeng, Y.M.; Wang, T.L.; Shih Ie, M. Mutation and loss of expression of ARID1A in uterine low-grade endometrioid carcinoma. Am. J. Surg. Pathol. 2011, 35, 625–632. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhao, H.; Zhang, X.; Wood, L.D.; Anders, R.A.; Choti, M.A.; Pawlik, T.M.; Daniel, H.D.; Kannangai, R.; Offerhaus, G.J.; et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat. Genet. 2011, 43, 828–829. [Google Scholar] [CrossRef] [Green Version]
- Drost, J.; Mantovani, F.; Tocco, F.; Elkon, R.; Comel, A.; Holstege, H.; Kerkhoven, R.; Jonkers, J.; Voorhoeve, P.M.; Agami, R.; et al. BRD7 is a candidate tumour suppressor gene required for p53 function. Nat. Cell Biol. 2010, 12, 380–389. [Google Scholar] [CrossRef]
- Sun, A.; Tawfik, O.; Gayed, B.; Thrasher, J.B.; Hoestje, S.; Li, C.; Li, B. Aberrant expression of SWI/SNF catalytic subunits BRG1/BRM is associated with tumor development and increased invasiveness in prostate cancers. Prostate 2007, 67, 203–213. [Google Scholar] [CrossRef] [PubMed]
- De Zwaan, S.E.; Haass, N.K. Genetics of basal cell carcinoma. Australas. J. Dermatol. 2010, 51, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, A.; Papazoglu, C.; Wu, Y.; Capurso, D.; Brodt, M.; Francis, D.; Bredel, M.; Vogel, H.; Mills, A.A. CHD5 is a tumor suppressor at human 1p36. Cell 2007, 128, 459–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Chung, N.G.; Kang, M.R.; Yoo, N.J.; Lee, S.H. Genetic and expressional alterations of CHD genes in gastric and colorectal cancers. Histopathology 2011, 58, 660–668. [Google Scholar] [CrossRef]
- Wang, J.; Chen, H.; Fu, S.; Xu, Z.M.; Sun, K.L.; Fu, W.N. The involvement of CHD5 hypermethylation in laryngeal squamous cell carcinoma. Oral. Oncol. 2011, 47, 601–608. [Google Scholar] [CrossRef]
- Wessels, K.; Bohnhorst, B.; Luhmer, I.; Morlot, S.; Bohring, A.; Jonasson, J.; Epplen, J.T.; Gadzicki, D.; Glaser, S.; Gohring, G.; et al. Novel CHD7 mutations contributing to the mutation spectrum in patients with CHARGE syndrome. Eur. J. Med. Genet. 2010, 53, 280–285. [Google Scholar] [CrossRef]
- Mattera, L.; Escaffit, F.; Pillaire, M.J.; Selves, J.; Tyteca, S.; Hoffmann, J.S.; Gourraud, P.A.; Chevillard-Briet, M.; Cazaux, C.; Trouche, D. The p400/Tip60 ratio is critical for colorectal cancer cell proliferation through DNA damage response pathways. Oncogene 2009, 28, 1506–1517. [Google Scholar] [CrossRef] [Green Version]
- Varela, I.; Tarpey, P.; Raine, K.; Huang, D.; Ong, C.K.; Stephens, P.; Davies, H.; Jones, D.; Lin, M.L.; Teague, J.; et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011, 469, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef]
- Balakrishnan, A.; Bleeker, F.E.; Lamba, S.; Rodolfo, M.; Daniotti, M.; Scarpa, A.; van Tilborg, A.A.; Leenstra, S.; Zanon, C.; Bardelli, A. Novel somatic and germline mutations in cancer candidate genes in glioblastoma, melanoma, and pancreatic carcinoma. Cancer Res. 2007, 67, 3545–3550. [Google Scholar] [CrossRef] [Green Version]
- Morrison, D.K. MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304. [Google Scholar] [CrossRef] [Green Version]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Yang, J. Functional mechanisms for human tumor suppressors. J. Cancer 2010, 1, 136–140. [Google Scholar] [CrossRef]
- Shortt, J.; Johnstone, R.W. Oncogenes in cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012, 4, a009829. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Rodenhuis, S. Ras and human tumors. Semin. Cancer Biol. 1992, 3, 241–247. [Google Scholar] [PubMed]
- Brison, O. Gene amplification and tumor progression. Biochim. Biophys. Acta 1993, 1155, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Alitalo, K.; Klempnauer, K.H.; Varmus, H.E.; Bishop, J.M.; Gilbert, F.; Brodeur, G.; Goldstein, M.; Trent, J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983, 305, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Kruidering, M. N-MYC overexpression and neuroblastomas. Trends Pharmacol. Sci. 2002, 23, 452. [Google Scholar] [CrossRef] [PubMed]
- Zajac-Kaye, M. Myc oncogene: A key component in cell cycle regulation and its implication for lung cancer. Lung Cancer 2001, 34 (Suppl. 2), S43–S46. [Google Scholar] [CrossRef]
- Lane, D.P. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [Green Version]
- Li, F.P.; Fraumeni, J.F., Jr.; Mulvihill, J.J.; Blattner, W.A.; Dreyfus, M.G.; Tucker, M.A.; Miller, R.W. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988, 48, 5358–5362. [Google Scholar]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F., Jr.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A.; et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef]
- You, J.S.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatziapostolou, M.; Iliopoulos, D. Epigenetic aberrations during oncogenesis. Cell. Mol. Life Sci. 2011, 68, 1681–1702. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7, 823–833. [Google Scholar] [CrossRef]
- Hawkins, R.D.; Hon, G.C.; Yang, C.; Antosiewicz-Bourget, J.E.; Lee, L.K.; Ngo, Q.M.; Klugman, S.; Ching, K.A.; Edsall, L.E.; Ye, Z.; et al. Dynamic chromatin states in human ES cells reveal potential regulatory sequences and genes involved in pluripotency. Cell Res. 2011, 21, 1393–1409. [Google Scholar] [CrossRef] [Green Version]
- Hon, G.C.; Hawkins, R.D.; Ren, B. Predictive chromatin signatures in the mammalian genome. Hum. Mol. Genet. 2009, 18, R195–R201. [Google Scholar] [CrossRef] [Green Version]
- Mills, A.A. Throwing the cancer switch: Reciprocal roles of polycomb and trithorax proteins. Nat. Rev. Cancer 2010, 10, 669–682. [Google Scholar] [CrossRef]
- Lin, H.; Wong, R.P.; Martinka, M.; Li, G. Loss of SNF5 expression correlates with poor patient survival in melanoma. Clin. Cancer Res. 2009, 15, 6404–6411. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, Y.; Yang, Y.; Niu, M.; Sun, S.; Ji, H.; Ma, Y.; Yao, G.; Jiang, Y.; Shan, M.; et al. Frequent low expression of chromatin remodeling gene ARID1A in breast cancer and its clinical significance. Cancer Epidemiol. 2012, 36, 288–293. [Google Scholar] [CrossRef]
- MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, A.; Rathmell, J.C. Metabolic Barriers to T Cell Function in Tumors. J. Immunol. 2018, 200, 400–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottensmeier, C.H.; Perry, K.L.; Harden, E.L.; Stasakova, J.; Jenei, V.; Fleming, J.; Wood, O.; Woo, J.; Woelk, C.H.; Thomas, G.J.; et al. Upregulated Glucose Metabolism Correlates Inversely with CD8+ T-cell Infiltration and Survival in Squamous Cell Carcinoma. Cancer Res. 2016, 76, 4136–4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis-Yadley, A.H.; Abbott, A.M.; Pimiento, J.M.; Chen, D.T.; Malafa, M.P. Increased Expression of the Glucose Transporter Type 1 Gene Is Associated With Worse Overall Survival in Resected Pancreatic Adenocarcinoma. Pancreas 2016, 45, 974–979. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; Deoliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Andrejeva, G.; Rathmell, J.C. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017, 26, 49–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hardie, R.A.; Hoy, A.J.; van Geldermalsen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015, 236, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Mocellin, S.; Bronte, V.; Nitti, D. Nitric oxide, a double edged sword in cancer biology: Searching for therapeutic opportunities. Med. Res. Rev. 2007, 27, 317–352. [Google Scholar] [CrossRef]
- Grzywa, T.M.; Sosnowska, A.; Matryba, P.; Rydzynska, Z.; Jasinski, M.; Nowis, D.; Golab, J. Myeloid Cell-Derived Arginase in Cancer Immune Response. Front. Immunol. 2020, 11, 938. [Google Scholar] [CrossRef] [PubMed]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brody, J.R.; Costantino, C.L.; Berger, A.C.; Sato, T.; Lisanti, M.P.; Yeo, C.J.; Emmons, R.V.; Witkiewicz, A.K. Expression of indoleamine 2,3-dioxygenase in metastatic malignant melanoma recruits regulatory T cells to avoid immune detection and affects survival. Cell Cycle 2009, 8, 1930–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witkiewicz, A.K.; Costantino, C.L.; Metz, R.; Muller, A.J.; Prendergast, G.C.; Yeo, C.J.; Brody, J.R. Genotyping and expression analysis of IDO2 in human pancreatic cancer: A novel, active target. J. Am. Coll. Surg. 2009, 208, 781–787; discussion 787–789. [Google Scholar] [CrossRef] [Green Version]
- Inaba, T.; Ino, K.; Kajiyama, H.; Yamamoto, E.; Shibata, K.; Nawa, A.; Nagasaka, T.; Akimoto, H.; Takikawa, O.; Kikkawa, F. Role of the immunosuppressive enzyme indoleamine 2,3-dioxygenase in the progression of ovarian carcinoma. Gynecol. Oncol. 2009, 115, 185–192. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; You, S.; McGrath, B.C.; Cavener, D.R.; Vacca, C.; Orabona, C.; Bianchi, R.; Belladonna, M.L.; Volpi, C.; et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J. Immunol. 2006, 176, 6752–6761. [Google Scholar] [CrossRef] [Green Version]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Shoshan, J.; Maysel-Auslender, S.; Mor, A.; Keren, G.; George, J. Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1alpha. Eur. J. Immunol. 2008, 38, 2412–2418. [Google Scholar] [CrossRef] [PubMed]
- Clambey, E.T.; McNamee, E.N.; Westrich, J.A.; Glover, L.E.; Campbell, E.L.; Jedlicka, P.; de Zoeten, E.F.; Cambier, J.C.; Stenmark, K.R.; Colgan, S.P.; et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. USA 2012, 109, E2784–E2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, B.R.; Vignali, P.D.A.; Rittenhouse, N.L.; Scharping, N.E.; Peralta, R.; Lontos, K.; Frisch, A.T.; Delgoffe, G.M.; Poholek, A.C. Tumor microenvironmental signals reshape chromatin landscapes to limit the functional potential of exhausted T cells. Sci. Immunol. 2022, 7, eabj9123. [Google Scholar] [CrossRef]
- Vignali, P.D.A.; DePeaux, K.; Watson, M.J.; Ye, C.; Ford, B.R.; Lontos, K.; McGaa, N.K.; Scharping, N.E.; Menk, A.V.; Robson, S.C.; et al. Hypoxia drives CD39-dependent suppressor function in exhausted T cells to limit antitumor immunity. Nat. Immunol. 2023, 24, 267–279. [Google Scholar] [CrossRef]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1 alpha., and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [Green Version]
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014, 74, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Conforti, L. Potassium channels of T lymphocytes take center stage in the fight against cancer. J. Immunother. Cancer 2017, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Eil, R.; Vodnala, S.K.; Clever, D.; Klebanoff, C.A.; Sukumar, M.; Pan, J.H.; Palmer, D.C.; Gros, A.; Yamamoto, T.N.; Patel, S.J.; et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature 2016, 537, 539–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baixauli, F.; Villa, M.; Pearce, E.L. Potassium shapes antitumor immunity. Science 2019, 363, 1395–1396. [Google Scholar] [CrossRef] [PubMed]
- Vodnala, S.K.; Eil, R.; Kishton, R.J.; Sukumar, M.; Yamamoto, T.N.; Ha, N.H.; Lee, P.H.; Shin, M.; Patel, S.J.; Yu, Z.; et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 2019, 363, eaau0135. [Google Scholar] [CrossRef] [PubMed]
- Chimote, A.A.; Balajthy, A.; Arnold, M.J.; Newton, H.S.; Hajdu, P.; Qualtieri, J.; Wise-Draper, T.; Conforti, L. A defect in KCa3.1 channel activity limits the ability of CD8(+) T cells from cancer patients to infiltrate an adenosine-rich microenvironment. Sci. Signal 2018, 11, eaaq1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrie, E.; Coronas, V.; Constantin, B. Role of the calcium toolkit in cancer stem cells. Cell Calcium 2019, 80, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Immler, R.; Simon, S.I.; Sperandio, M. Calcium signalling and related ion channels in neutrophil recruitment and function. Eur. J. Clin. Investig. 2018, 48 (Suppl. 2), e12964. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnoczky, G. Intracellular Ca(2+) Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Bauer, C.A.; Kim, E.Y.; Marangoni, F.; Carrizosa, E.; Claudio, N.M.; Mempel, T.R. Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J. Clin. Investig. 2014, 124, 2425–2440. [Google Scholar] [CrossRef]
- Marangoni, F.; Murooka, T.T.; Manzo, T.; Kim, E.Y.; Carrizosa, E.; Elpek, N.M.; Mempel, T.R. The transcription factor NFAT exhibits signal memory during serial T cell interactions with antigen-presenting cells. Immunity 2013, 38, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Stewart, T.A.; Yapa, K.T.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta 2015, 1848, 2502–2511. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.D.; Bae, S.; Capece, T.; Nedelkovska, H.; de Rubio, R.G.; Smrcka, A.V.; Jun, C.D.; Jung, W.; Park, B.; Kim, T.I.; et al. Targeted calcium influx boosts cytotoxic T lymphocyte function in the tumour microenvironment. Nat. Commun. 2017, 8, 15365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarosz, E.L.; Chang, C.H. The Role of Reactive Oxygen Species in Regulating T Cell-mediated Immunity and Disease. Immune Netw. 2018, 18, e14. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, M.; Zhang, B.; Zhang, Y. Reactive Oxygen Species Regulate T Cell Immune Response in the Tumor Microenvironment. Oxid. Med. Cell. Longev. 2016, 2016, 1580967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franchina, D.G.; Dostert, C.; Brenner, D. Reactive Oxygen Species: Involvement in T Cell Signaling and Metabolism. Trends Immunol. 2018, 39, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusmartsev, S.; Nefedova, Y.; Yoder, D.; Gabrilovich, D.I. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 2004, 172, 989–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmielau, J.; Finn, O.J. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001, 61, 4756–4760. [Google Scholar] [PubMed]
- Ma, X.; Bi, E.; Lu, Y.; Su, P.; Huang, C.; Liu, L.; Wang, Q.; Yang, M.; Kalady, M.F.; Qian, J.; et al. Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab. 2019, 30, 143–156. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.L.; Lucas, J.E.; Schroeder, T.; Mori, S.; Wu, J.; Nevins, J.; Dewhirst, M.; West, M.; Chi, J.T. The genomic analysis of lactic acidosis and acidosis response in human cancers. PLoS Genet. 2008, 4, e1000293. [Google Scholar] [CrossRef] [Green Version]
- Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelin, A.; Gil-de-Gomez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., 3rd; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laoui, D.; Movahedi, K.; Van Overmeire, E.; Van den Bossche, J.; Schouppe, E.; Mommer, C.; Nikolaou, A.; Morias, Y.; De Baetselier, P.; Van Ginderachter, J.A. Tumor-associated macrophages in breast cancer: Distinct subsets, distinct functions. Int. J. Dev. Biol. 2011, 55, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian Res. 2014, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Li, C.; Xu, X.; Wei, S.; Jiang, P.; Xue, L.; Wang, J.; Senior, C. Tumor-associated macrophages: Potential therapeutic strategies and future prospects in cancer. J. Immunother. Cancer 2021, 9, e001341. [Google Scholar] [CrossRef]
- Gazzaniga, S.; Bravo, A.I.; Guglielmotti, A.; van Rooijen, N.; Maschi, F.; Vecchi, A.; Mantovani, A.; Mordoh, J.; Wainstok, R. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J. Investig. Dermatol. 2007, 127, 2031–2041. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.Y.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.; Zhou, Y.; Bu, H.; Lv, T.; Shi, Y.; Yang, J. Deletion of interleukin-6 in monocytes/macrophages suppresses the initiation of hepatocellular carcinoma in mice. J. Exp. Clin. Cancer Res. 2016, 35, 131. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.T.; Dai, Z.; Song, K.; Zhang, Z.J.; Zhou, Z.J.; Zhou, S.L.; Zhao, Y.M.; Xiao, Y.S.; Sun, Q.M.; Ding, Z.B.; et al. Macrophage-secreted IL-8 induces epithelial-mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int. J. Oncol. 2015, 46, 587–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Deng, J.; Rychahou, P.G.; Qiu, S.; Evers, B.M.; Zhou, B.P. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell 2009, 15, 416–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawata, M.; Koinuma, D.; Ogami, T.; Umezawa, K.; Iwata, C.; Watabe, T.; Miyazono, K. TGF-beta-induced epithelial-mesenchymal transition of A549 lung adenocarcinoma cells is enhanced by pro-inflammatory cytokines derived from RAW 264.7 macrophage cells. J. Biochem. 2012, 151, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasiljeva, O.; Papazoglou, A.; Kruger, A.; Brodoefel, H.; Korovin, M.; Deussing, J.; Augustin, N.; Nielsen, B.S.; Almholt, K.; Bogyo, M.; et al. Tumor cell-derived and macrophage-derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Res. 2006, 66, 5242–5250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gocheva, V.; Wang, H.W.; Gadea, B.B.; Shree, T.; Hunter, K.E.; Garfall, A.L.; Berman, T.; Joyce, J.A. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010, 24, 241–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, E.Y.; Li, J.F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.N.; Pollard, J.W. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.Y.; Pollard, J.W. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. 2007, 67, 5064–5066. [Google Scholar] [CrossRef] [Green Version]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef]
- Giraudo, E.; Inoue, M.; Hanahan, D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Investig. 2004, 114, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Leek, R.D.; Harris, A.L.; Lewis, C.E. Cytokine networks in solid human tumors: Regulation of angiogenesis. J. Leukoc. Biol. 1994, 56, 423–435. [Google Scholar] [CrossRef]
- Gil-Bernabe, A.M.; Ferjancic, S.; Tlalka, M.; Zhao, L.; Allen, P.D.; Im, J.H.; Watson, K.; Hill, S.A.; Amirkhosravi, A.; Francis, J.L.; et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood 2012, 119, 3164–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Zhang, X.H.; Massague, J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell 2011, 20, 538–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Mu, E.; Wei, Y.; Riethdorf, S.; Yang, Q.; Yuan, M.; Yan, J.; Hua, Y.; Tiede, B.J.; Lu, X.; et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell 2011, 20, 701–714. [Google Scholar] [CrossRef] [Green Version]
- Koukourakis, M.I.; Giatromanolaki, A.; Kakolyris, S.; O’Byrne, K.J.; Apostolikas, N.; Skarlatos, J.; Gatter, K.C.; Harris, A.L. Different patterns of stromal and cancer cell thymidine phosphorylase reactivity in non-small-cell lung cancer: Impact on tumour neoangiogenesis and survival. Br. J. Cancer 1998, 77, 1696–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, M.L.; Jones, N.R.; Candy, E.; Nicoll, J.A.; Compton, J.S.; Hughes, J.T.; Esiri, M.M.; Moss, T.H.; Cruz-Sanchez, F.F.; Coakham, H.B. The mononuclear cell infiltrate compared with survival in high-grade astrocytomas. Acta Neuropathol. 1989, 78, 189–193. [Google Scholar] [CrossRef]
- Shimura, S.; Yang, G.; Ebara, S.; Wheeler, T.M.; Frolov, A.; Thompson, T.C. Reduced infiltration of tumor-associated macrophages in human prostate cancer: Association with cancer progression. Cancer Res. 2000, 60, 5857–5861. [Google Scholar]
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Yang, L.; Yue, D.; Cao, L.; Li, L.; Wang, D.; Ping, Y.; Shen, Z.; Zheng, Y.; Wang, L.; et al. Macrophage-derived CCL22 promotes an immunosuppressive tumor microenvironment via IL-8 in malignant pleural effusion. Cancer Lett. 2019, 452, 244–253. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Ping, Q.; Yan, R.; Cheng, X.; Wang, W.; Zhong, Y.; Hou, Z.; Shi, Y.; Wang, C.; Li, R. Cancer-associated fibroblasts: Overview, progress, challenges, and directions. Cancer Gene Ther. 2021, 28, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, E.V.; Pereira, B.A.; Lawrence, M.G.; Ma, X.; Rebello, R.J.; Chan, H.; Niranjan, B.; Wu, Y.; Ellem, S.; Guan, X.; et al. Proteomic Profiling of Human Prostate Cancer-associated Fibroblasts (CAF) Reveals LOXL2-dependent Regulation of the Tumor Microenvironment. Mol. Cell Proteom. 2019, 18, 1410–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Hou, Y.; Yang, G.; Wang, X.; Tang, S.; Du, Y.E.; Yang, L.; Yu, T.; Zhang, H.; Zhou, M.; et al. Stromal miR-200s contribute to breast cancer cell invasion through CAF activation and ECM remodeling. Cell Death Differ. 2016, 23, 132–145. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, H.; Sahai, E. Mechanisms and impact of altered tumour mechanics. Nat. Cell Biol. 2018, 20, 766–774. [Google Scholar] [CrossRef]
- Jain, R.K.; Martin, J.D.; Stylianopoulos, T. The role of mechanical forces in tumor growth and therapy. Annu. Rev. Biomed. Eng. 2014, 16, 321–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DuFort, C.C.; DelGiorno, K.E.; Hingorani, S.R. Mounting Pressure in the Microenvironment: Fluids, Solids, and Cells in Pancreatic Ductal Adenocarcinoma. Gastroenterology 2016, 150, 1545–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [Green Version]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [Green Version]
- Dumont, N.; Liu, B.; Defilippis, R.A.; Chang, H.; Rabban, J.T.; Karnezis, A.N.; Tjoe, J.A.; Marx, J.; Parvin, B.; Tlsty, T.D. Breast fibroblasts modulate early dissemination, tumorigenesis, and metastasis through alteration of extracellular matrix characteristics. Neoplasia 2013, 15, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Madsen, C.D.; Pedersen, J.T.; Venning, F.A.; Singh, L.B.; Moeendarbary, E.; Charras, G.; Cox, T.R.; Sahai, E.; Erler, J.T. Hypoxia and loss of PHD2 inactivate stromal fibroblasts to decrease tumour stiffness and metastasis. EMBO Rep. 2015, 16, 1394–1408. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Cespedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Malanchi, I.; Santamaria-Martinez, A.; Susanto, E.; Peng, H.; Lehr, H.A.; Delaloye, J.F.; Huelsken, J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2011, 481, 85–89. [Google Scholar] [CrossRef] [Green Version]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massague, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wever, O.; Nguyen, Q.D.; Van Hoorde, L.; Bracke, M.; Bruyneel, E.; Gespach, C.; Mareel, M. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004, 18, 1016–1018. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef]
- Cazet, A.S.; Hui, M.N.; Elsworth, B.L.; Wu, S.Z.; Roden, D.; Chan, C.L.; Skhinas, J.N.; Collot, R.; Yang, J.; Harvey, K.; et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat. Commun. 2018, 9, 2897. [Google Scholar] [CrossRef] [Green Version]
- Bruzzese, F.; Hagglof, C.; Leone, A.; Sjoberg, E.; Roca, M.S.; Kiflemariam, S.; Sjoblom, T.; Hammarsten, P.; Egevad, L.; Bergh, A.; et al. Local and systemic protumorigenic effects of cancer-associated fibroblast-derived GDF15. Cancer Res. 2014, 74, 3408–3417. [Google Scholar] [CrossRef] [Green Version]
- Olumi, A.F.; Grossfeld, G.D.; Hayward, S.W.; Carroll, P.R.; Tlsty, T.D.; Cunha, G.R. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999, 59, 5002–5011. [Google Scholar] [CrossRef] [PubMed]
- Bhome, R.; Goh, R.W.; Bullock, M.D.; Pillar, N.; Thirdborough, S.M.; Mellone, M.; Mirnezami, R.; Galea, D.; Veselkov, K.; Gu, Q.; et al. Exosomal microRNAs derived from colorectal cancer-associated fibroblasts: Role in driving cancer progression. Aging 2017, 9, 2666–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 2012, 151, 1542–1556. [Google Scholar] [CrossRef] [Green Version]
- Fearon, D.T. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol. Res. 2014, 2, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakins, M.A.; Ghorani, E.; Munir, H.; Martins, C.P.; Shields, J.D. Cancer-associated fibroblasts induce antigen-specific deletion of CD8(+) T Cells to protect tumour cells. Nat. Commun. 2018, 9, 948. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinchuk, I.V.; Saada, J.I.; Beswick, E.J.; Boya, G.; Qiu, S.M.; Mifflin, R.C.; Raju, G.S.; Reyes, V.E.; Powell, D.W. PD-1 ligand expression by human colonic myofibroblasts/fibroblasts regulates CD4+ T-cell activity. Gastroenterology 2008, 135, 1228–1237. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Albelda, S.M. Tumor-associated neutrophils: Friend or foe? Carcinogenesis 2012, 33, 949–955. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Swierczak, A.; Mouchemore, K.A.; Hamilton, J.A.; Anderson, R.L. Neutrophils: Important contributors to tumor progression and metastasis. Cancer Metastasis Rev. 2015, 34, 735–751. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A. The yin-yang of tumor-associated neutrophils. Cancer Cell 2009, 16, 173–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uribe-Querol, E.; Rosales, C. Neutrophils in Cancer: Two Sides of the Same Coin. J. Immunol. Res. 2015, 2015, 983698. [Google Scholar] [CrossRef] [Green Version]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limon, P. The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eruslanov, E.B.; Singhal, S.; Albelda, S.M. Mouse versus Human Neutrophils in Cancer: A Major Knowledge Gap. Trends Cancer 2017, 3, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Taya, M.; Garcia-Hernandez, M.L.; Rangel-Moreno, J.; Minor, B.; Gibbons, E.; Hammes, S.R. Neutrophil elastase from myeloid cells promotes TSC2-null tumor growth. Endocr. Relat. Cancer 2020, 27, 261–274. [Google Scholar] [CrossRef]
- Lerman, I.; Garcia-Hernandez, M.L.; Rangel-Moreno, J.; Chiriboga, L.; Pan, C.; Nastiuk, K.L.; Krolewski, J.J.; Sen, A.; Hammes, S.R. Infiltrating Myeloid Cells Exert Protumorigenic Actions via Neutrophil Elastase. Mol. Cancer Res. 2017, 15, 1138–1152. [Google Scholar] [CrossRef] [Green Version]
- Liao, Z.; Chua, D.; Tan, N.S. Reactive oxygen species: A volatile driver of field cancerization and metastasis. Mol. Cancer 2019, 18, 65. [Google Scholar] [CrossRef]
- Wu, L.; Saxena, S.; Singh, R.K. Neutrophils in the Tumor Microenvironment. In Tumor Microenvironment; Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2020; Volume 1224, pp. 1–20. [Google Scholar] [CrossRef]
- Coussens, L.M.; Tinkle, C.L.; Hanahan, D.; Werb, Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell 2000, 103, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Masucci, M.T.; Minopoli, M.; Del Vecchio, S.; Carriero, M.V. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front. Immunol. 2020, 11, 1749. [Google Scholar] [CrossRef]
- Spicer, J.D.; McDonald, B.; Cools-Lartigue, J.J.; Chow, S.C.; Giannias, B.; Kubes, P.; Ferri, L.E. Neutrophils promote liver metastasis via Mac-1-mediated interactions with circulating tumor cells. Cancer Res. 2012, 72, 3919–3927. [Google Scholar] [CrossRef] [Green Version]
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Investig. 2013, 123, 3446–3458. [Google Scholar] [CrossRef] [PubMed]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, X.H. Tumor-Associated Neutrophils and Macrophages-Heterogenous but Not Chaotic. Front. Immunol. 2020, 11, 553967. [Google Scholar] [CrossRef]
- Itatani, Y.; Yamamoto, T.; Zhong, C.; Molinolo, A.A.; Ruppel, J.; Hegde, P.; Taketo, M.M.; Ferrara, N. Suppressing neutrophil-dependent angiogenesis abrogates resistance to anti-VEGF antibody in a genetic model of colorectal cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 21598–21608. [Google Scholar] [CrossRef] [PubMed]
- Grosse-Steffen, T.; Giese, T.; Giese, N.; Longerich, T.; Schirmacher, P.; Hansch, G.M.; Gaida, M.M. Epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma and pancreatic tumor cell lines: The role of neutrophils and neutrophil-derived elastase. Clin. Dev. Immunol. 2012, 2012, 720768. [Google Scholar] [CrossRef]
- Li, S.; Cong, X.; Gao, H.; Lan, X.; Li, Z.; Wang, W.; Song, S.; Wang, Y.; Li, C.; Zhang, H.; et al. Tumor-associated neutrophils induce EMT by IL-17a to promote migration and invasion in gastric cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Mollinedo, F. Neutrophil Degranulation, Plasticity, and Cancer Metastasis. Trends Immunol. 2019, 40, 228–242. [Google Scholar] [CrossRef]
- Mishalian, I.; Bayuh, R.; Eruslanov, E.; Michaeli, J.; Levy, L.; Zolotarov, L.; Singhal, S.; Albelda, S.M.; Granot, Z.; Fridlender, Z.G. Neutrophils recruit regulatory T-cells into tumors via secretion of CCL17—A new mechanism of impaired antitumor immunity. Int. J. Cancer 2014, 135, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Raftopoulou, S.; Valadez-Cosmes, P.; Mihalic, Z.N.; Schicho, R.; Kargl, J. Tumor-Mediated Neutrophil Polarization and Therapeutic Implications. Int. J. Mol. Sci. 2022, 23, 3218. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Hyman, D.M.; Davids, M.S.; Siu, L.L. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet 2020, 395, 1078–1088. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal. Transduct. Target Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Lito, P.; Solomon, M.; Li, L.S.; Hansen, R.; Rosen, N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016, 351, 604–608. [Google Scholar] [CrossRef] [Green Version]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589. [Google Scholar] [CrossRef] [Green Version]
- Zeng, M.; Lu, J.; Li, L.; Feru, F.; Quan, C.; Gero, T.W.; Ficarro, S.B.; Xiong, Y.; Ambrogio, C.; Paranal, R.M.; et al. Potent and Selective Covalent Quinazoline Inhibitors of KRAS G12C. Cell Chem. Biol. 2017, 24, 1005–1016. [Google Scholar] [CrossRef] [Green Version]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Pietras, R.J.; Fendly, B.M.; Chazin, V.R.; Pegram, M.D.; Howell, S.B.; Slamon, D.J. Antibody to HER-2/neu receptor blocks DNA repair after cisplatin in human breast and ovarian cancer cells. Oncogene 1994, 9, 1829–1838. [Google Scholar]
- Izumi, Y.; Xu, L.; di Tomaso, E.; Fukumura, D.; Jain, R.K. Tumour biology: Herceptin acts as an anti-angiogenic cocktail. Nature 2002, 416, 279–280. [Google Scholar] [CrossRef]
- Harding, J.; Burtness, B. Cetuximab: An epidermal growth factor receptor chemeric human-murine monoclonal antibody. Drugs Today 2005, 41, 107–127. [Google Scholar] [CrossRef]
- Maier, L.A.; Xu, F.J.; Hester, S.; Boyer, C.M.; McKenzie, S.; Bruskin, A.M.; Argon, Y.; Bast, R.C., Jr. Requirements for the internalization of a murine monoclonal antibody directed against the HER-2/neu gene product c-erbB-2. Cancer Res. 1991, 51, 5361–5369. [Google Scholar] [PubMed]
- Redman, J.M.; Hill, E.M.; AlDeghaither, D.; Weiner, L.M. Mechanisms of action of therapeutic antibodies for cancer. Mol. Immunol. 2015, 67, 28–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Coffin, R. Interview with Robert Coffin, inventor of T-VEC: The first oncolytic immunotherapy approved for the treatment of cancer. Immunotherapy 2016, 8, 103–106. [Google Scholar] [CrossRef] [Green Version]
- Azangou-Khyavy, M.; Ghasemi, M.; Khanali, J.; Boroomand-Saboor, M.; Jamalkhah, M.; Soleimani, M.; Kiani, J. CRISPR/Cas: From Tumor Gene Editing to T Cell-Based Immunotherapy of Cancer. Front. Immunol. 2020, 11, 2062. [Google Scholar] [CrossRef]
- Chira, S.; Gulei, D.; Hajitou, A.; Berindan-Neagoe, I. Restoring the p53 ‘Guardian’ Phenotype in p53-Deficient Tumor Cells with CRISPR/Cas9. Trends Biotechnol. 2018, 36, 653–660. [Google Scholar] [CrossRef]
- Wang, H.; Sun, W. CRISPR-mediated targeting of HER2 inhibits cell proliferation through a dominant negative mutation. Cancer Lett. 2017, 385, 137–143. [Google Scholar] [CrossRef]
- Su, S.; Hu, B.; Shao, J.; Shen, B.; Du, J.; Du, Y.; Zhou, J.; Yu, L.; Zhang, L.; Chen, F.; et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci. Rep. 2016, 6, 20070. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Zou, Y.; Zhang, L.; Tang, J.; Niedermann, G.; Firat, E.; Huang, X.; Zhu, X. Nucleofection with Plasmid DNA for CRISPR/Cas9-Mediated Inactivation of Programmed Cell Death Protein 1 in CD133-Specific CAR T Cells. Hum. Gene Ther. 2019, 30, 446–458. [Google Scholar] [CrossRef]
- Dai, X.; Park, J.J.; Du, Y.; Kim, H.R.; Wang, G.; Errami, Y.; Chen, S. One-step generation of modular CAR-T cells with AAV-Cpf1. Nat. Methods 2019, 16, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gonen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell, K.J.; Gottschalk, S.; Talleur, A.C. Allogeneic CAR Cell Therapy-More Than a Pipe Dream. Front. Immunol. 2020, 11, 618427. [Google Scholar] [CrossRef] [PubMed]
- Pavel-Dinu, M.; Wiebking, V.; Dejene, B.T.; Srifa, W.; Mantri, S.; Nicolas, C.E.; Lee, C.; Bao, G.; Kildebeck, E.J.; Punjya, N.; et al. Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat. Commun. 2019, 10, 1634. [Google Scholar] [CrossRef] [Green Version]
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, L.H. Cancer vaccines. BMJ 2015, 350, h988. [Google Scholar] [CrossRef]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Lower, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrors, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbelaez, C.A.; Estrada, J.; Gessner, M.A.; Glaus, C.; Morales, A.B.; Mohn, D.; Phee, H.; Lipford, J.R.; Johnston, J.A. A nanoparticle vaccine that targets neoantigen peptides to lymphoid tissues elicits robust antitumor T cell responses. NPJ Vaccines 2020, 5, 106. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug. Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Cafri, G.; Gartner, J.J.; Zaks, T.; Hopson, K.; Levin, N.; Paria, B.C.; Parkhurst, M.R.; Yossef, R.; Lowery, F.J.; Jafferji, M.S.; et al. mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J. Clin. Investig. 2020, 130, 5976–5988. [Google Scholar] [CrossRef]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2017, 38, 226–256. [Google Scholar] [CrossRef]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005, 10, 86–103. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Gong, J. Heat Shock Proteins Promote Cancer: It’s a Protection Racket. Trends Biochem. Sci. 2016, 41, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.F.; Ren, W.; Rollins, L.; Pittman, P.; Shah, M.; Shen, L.; Gu, Q.; Strube, R.; Hu, F.; Chen, S.Y. A broadly applicable, personalized heat shock protein-mediated oncolytic tumor vaccine. Cancer Res. 2003, 63, 7321–7329. [Google Scholar]
- Taha, E.A.; Ono, K.; Eguchi, T. Roles of Extracellular HSPs as Biomarkers in Immune Surveillance and Immune Evasion. Int. J. Mol. Sci. 2019, 20, 4588. [Google Scholar] [CrossRef] [Green Version]
- Murshid, A.; Gong, J.; Stevenson, M.A.; Calderwood, S.K. Heat shock proteins and cancer vaccines: Developments in the past decade and chaperoning in the decade to come. Expert Rev. Vaccines 2011, 10, 1553–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, J.K.; Xiong, X.; Ren, X.; Yang, J.M.; Song, J. Heat Shock Proteins in Cancer Immunotherapy. J. Oncol. 2019, 2019, 3267207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, G.; Tamura, Y.; Hirai, I.; Kamiguchi, K.; Ichimiya, S.; Torigoe, T.; Hiratsuka, H.; Sunakawa, H.; Sato, N. Tumor-derived heat shock protein 70-pulsed dendritic cells elicit tumor-specific cytotoxic T lymphocytes (CTLs) and tumor immunity. Cancer Sci. 2004, 95, 248–253. [Google Scholar] [CrossRef]
- Hoos, A.; Levey, D.L. Vaccination with heat shock protein-peptide complexes: From basic science to clinical applications. Expert. Rev. Vaccines 2003, 2, 369–379. [Google Scholar] [CrossRef]
- Blachere, N.E.; Udono, H.; Janetzki, S.; Li, Z.; Heike, M.; Srivastava, P.K. Heat shock protein vaccines against cancer. J. Immunother. Emphas. Tumor Immunol. 1993, 14, 352–356. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Y.; Xiong, W.; Yin, B.; Huang, Y.; Chu, J.; Xing, C.; Qian, C.; Du, Y.; Duan, T.; et al. Development of a TCR-like antibody and chimeric antigen receptor against NY-ESO-1/HLA-A2 for cancer immunotherapy. J. Immunother. Cancer 2022, 10, e004035. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Ivica, N.A.; Jia, B.; Li, Y.; Dong, H.; Liang, Y.; Brown, D.; Romee, R.; Chen, J. CAR-T cells targeting a nucleophosmin neoepitope exhibit potent specific activity in mouse models of acute myeloid leukaemia. Nat. Biomed. Eng. 2021, 5, 399–413. [Google Scholar] [CrossRef]
- Hsiue, E.H.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; DiNapoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a neoantigen derived from a common TP53 mutation. Science 2021, 371, eabc8697. [Google Scholar] [CrossRef]
- Douglass, J.; Hsiue, E.H.; Mog, B.J.; Hwang, M.S.; DiNapoli, S.R.; Pearlman, A.H.; Miller, M.S.; Wright, K.M.; Azurmendi, P.A.; Wang, Q.; et al. Bispecific antibodies targeting mutant RAS neoantigens. Sci. Immunol. 2021, 6, eabd5515. [Google Scholar] [CrossRef]
- Sergeeva, A.; He, H.; Ruisaard, K.; St John, L.; Alatrash, G.; Clise-Dwyer, K.; Li, D.; Patenia, R.; Hong, R.; Sukhumalchandra, P.; et al. Activity of 8F4, a T-cell receptor-like anti-PR1/HLA-A2 antibody, against primary human AML in vivo. Leukemia 2016, 30, 1475–1484. [Google Scholar] [CrossRef] [Green Version]
- Dao, T.; Yan, S.; Veomett, N.; Pankov, D.; Zhou, L.; Korontsvit, T.; Scott, A.; Whitten, J.; Maslak, P.; Casey, E.; et al. Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Sci. Transl. Med. 2013, 5, 176ra33. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Li, Y.M.; Zhou, J.J.; Zhou, Z.; Xu, Y.C.; Zhao, W.B.; Chen, S.Q. The Antitumor Activity of TCR-Mimic Antibody-Drug Conjugates (TCRm-ADCs) Targeting the Intracellular Wilms Tumor 1 (WT1) Oncoprotein. Int. J. Mol. Sci. 2019, 20, 3912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dao, T.; Mun, S.S.; Scott, A.C.; Jarvis, C.A.; Korontsvit, T.; Yang, Z.; Liu, L.; Klatt, M.G.; Guerreiro, M.; Selvakumar, A.; et al. Depleting T regulatory cells by targeting intracellular Foxp3 with a TCR mimic antibody. Oncoimmunology 2019, 8, 1570778. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.B.; Bivens, C.K.; Mobley, A.S.; Herrera, C.E.; McCormick, A.L.; Wichner, T.; Sabnani, M.K.; Wood, L.M.; Weidanz, J.A. TCR-like antibody drug conjugates mediate killing of tumor cells with low peptide/HLA targets. mAbs 2017, 9, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Garber, H.R.; Lu, S.; He, H.; Tallis, E.; Ding, X.; Sergeeva, A.; Wood, M.S.; Dotti, G.; Salvado, B.; et al. A novel TCR-like CAR with specificity for PR1/HLA-A2 effectively targets myeloid leukemia in vitro when expressed in human adult peripheral blood and cord blood T cells. Cytotherapy 2016, 18, 985–994. [Google Scholar] [CrossRef] [Green Version]
- Dao, T.; Pankov, D.; Scott, A.; Korontsvit, T.; Zakhaleva, V.; Xu, Y.; Xiang, J.; Yan, S.; de Morais Guerreiro, M.D.; Veomett, N.; et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat. Biotechnol. 2015, 33, 1079–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veomett, N.; Dao, T.; Liu, H.; Xiang, J.; Pankov, D.; Dubrovsky, L.; Whitten, J.A.; Park, S.M.; Korontsvit, T.; Zakhaleva, V.; et al. Therapeutic efficacy of an Fc-enhanced TCR-like antibody to the intracellular WT1 oncoprotein. Clin. Cancer Res. 2014, 20, 4036–4046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubrovsky, L.; Pankov, D.; Brea, E.J.; Dao, T.; Scott, A.; Yan, S.; O’Reilly, R.J.; Liu, C.; Scheinberg, D.A. A TCR-mimic antibody to WT1 bypasses tyrosine kinase inhibitor resistance in human BCR-ABL+ leukemias. Blood 2014, 123, 3296–3304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Kailayangiri, S.; Altvater, B.; Wiebel, M.; Jamitzky, S.; Rossig, C. Overcoming Heterogeneity of Antigen Expression for Effective CAR T Cell Targeting of Cancers. Cancers 2020, 12, 1075. [Google Scholar] [CrossRef]
- Anurathapan, U.; Chan, R.C.; Hindi, H.F.; Mucharla, R.; Bajgain, P.; Hayes, B.C.; Fisher, W.E.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; et al. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol. Ther. 2014, 22, 623–633. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishna, S.; Highfill, S.L.; Walsh, Z.; Nguyen, S.M.; Lei, H.; Shern, J.F.; Qin, H.; Kraft, I.L.; Stetler-Stevenson, M.; Yuan, C.M.; et al. Modulation of Target Antigen Density Improves CAR T-cell Functionality and Persistence. Clin. Cancer Res. 2019, 25, 5329–5341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pont, M.J.; Hill, T.; Cole, G.O.; Abbott, J.J.; Kelliher, J.; Salter, A.I.; Hudecek, M.; Comstock, M.L.; Rajan, A.; Patel, B.K.R.; et al. gamma-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019, 134, 1585–1597. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Zuo, S.; Deng, B.; Xu, X.; Li, C.; Zheng, Q.; Ling, Z.; Song, W.; Xu, J.; Duan, J.; et al. Sequential CD19-22 CAR T therapy induces sustained remission in children with r/r B-ALL. Blood 2020, 135, 387–391. [Google Scholar] [CrossRef]
- Wang, N.; Hu, X.; Cao, W.; Li, C.; Xiao, Y.; Cao, Y.; Gu, C.; Zhang, S.; Chen, L.; Cheng, J.; et al. Efficacy and safety of CAR19/22 T-cell cocktail therapy in patients with refractory/relapsed B-cell malignancies. Blood 2020, 135, 17–27. [Google Scholar] [CrossRef]
- Dai, H.; Wu, Z.; Jia, H.; Tong, C.; Guo, Y.; Ti, D.; Han, X.; Liu, Y.; Zhang, W.; Wang, C.; et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J. Hematol. Oncol. 2020, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schonfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther. Nucleic Acids 2013, 2, e105. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [Green Version]
- Schneider, D.; Xiong, Y.; Wu, D.; Nölle, V.; Schmitz, S.; Haso, W.; Kaiser, A.; Dropulic, B.; Orentas, R.J. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J. Immunother. Cancer 2017, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwahori, K.; Kakarla, S.; Velasquez, M.P.; Yu, F.; Yi, Z.; Gerken, C.; Song, X.T.; Gottschalk, S. Engager T cells: A new class of antigen-specific T cells that redirect bystander T cells. Mol. Ther. 2015, 23, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.G.; Marks, I.; Srinivasarao, M.; Kanduluru, A.K.; Mahalingam, S.M.; Liu, X.; Chu, H.; Low, P.S. Use of a Single CAR T Cell and Several Bispecific Adapters Facilitates Eradication of Multiple Antigenically Different Solid Tumors. Cancer Res. 2019, 79, 387–396. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, A.R.; Owens, M.N.; Geyer, C.R. Modular Chimeric Antigen Receptor Systems for Universal CAR T Cell Retargeting. Int. J. Mol. Sci. 2020, 21, 7222. [Google Scholar] [CrossRef] [PubMed]
- Urbanska, K.; Lanitis, E.; Poussin, M.; Lynn, R.C.; Gavin, B.P.; Kelderman, S.; Yu, J.; Scholler, N.; Powell, D.J., Jr. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 2012, 72, 1844–1852. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Ralpha2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Whilding, L.M.; Halim, L.; Draper, B.; Parente-Pereira, A.C.; Zabinski, T.; Davies, D.M.; Maher, J. CAR T-Cells Targeting the Integrin alphavbeta6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers 2019, 11, 674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, A.Y.; Na, M.; Dyson, K.A.; Grippin, A.J.; Deleyrolle, L.P.; Zhang, W.; et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 2019, 10, 4016. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Rui, W.; Zheng, H.; Huang, D.; Yu, F.; Zhang, Y.; Dong, J.; Zhao, X.; Lin, X. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur. J. Immunol. 2020, 50, 712–724. [Google Scholar] [CrossRef]
- Gregor, C.E.; Foeng, J.; Comerford, I.; McColl, S.R. Chemokine-Driven CD4(+) T Cell Homing: New Concepts and Recent Advances. Adv. Immunol. 2017, 135, 119–181. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, M.C.; Popplewell, L.; Cooper, L.J.; DiGiusto, D.; Kalos, M.; Ostberg, J.R.; Forman, S.J. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol. Blood Marrow Transplant. 2010, 16, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Lamers, C.H.; Gratama, J.W.; Pouw, N.M.; Langeveld, S.C.; Krimpen, B.A.; Kraan, J.; Stoter, G.; Debets, R. Parallel detection of transduced T lymphocytes after immunogene therapy of renal cell cancer by flow cytometry and real-time polymerase chain reaction: Implications for loss of transgene expression. Hum. Gene Ther. 2005, 16, 1452–1462. [Google Scholar] [CrossRef] [PubMed]
- Van der Stegen, S.J.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Zhang, M.; Ramos, C.A.; Durett, A.; Liu, E.; Dakhova, O.; Liu, H.; Creighton, C.J.; Gee, A.P.; Heslop, H.E.; et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 2014, 123, 3750–3759. [Google Scholar] [CrossRef] [Green Version]
- Sallusto, F.; Geginat, J.; Lanzavecchia, A. Central memory and effector memory T cell subsets: Function, generation, and maintenance. Annu. Rev. Immunol. 2004, 22, 745–763. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liot, S.; Balas, J.; Aubert, A.; Prigent, L.; Mercier-Gouy, P.; Verrier, B.; Bertolino, P.; Hennino, A.; Valcourt, U.; Lambert, E. Stroma Involvement in Pancreatic Ductal Adenocarcinoma: An Overview Focusing on Extracellular Matrix Proteins. Front. Immunol. 2021, 12, 612271. [Google Scholar] [CrossRef] [PubMed]
- Himes, B.T.; Geiger, P.A.; Ayasoufi, K.; Bhargav, A.G.; Brown, D.A.; Parney, I.F. Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front. Oncol. 2021, 11, 770561. [Google Scholar] [CrossRef]
- Walker, A.J.; Majzner, R.G.; Zhang, L.; Wanhainen, K.; Long, A.H.; Nguyen, S.M.; Lopomo, P.; Vigny, M.; Fry, T.J.; Orentas, R.J.; et al. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol. Ther. 2017, 25, 2189–2201. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Wang, Y.; Zou, Z.; Yang, Y.; Wang, X.; Xin, X.; Tu, S.; Li, Y. Combination strategies to optimize the efficacy of chimeric antigen receptor T cell therapy in haematological malignancies. Front. Immunol. 2022, 13, 954235. [Google Scholar] [CrossRef] [PubMed]
- Perica, K.; Flynn, J.; Curran, K.J.; Rivere, I.; Wang, X.; Senechal, B.; Halton, E.; Diamonte, C.; Pineda, J.; Bernal, Y.; et al. Impact of bridging chemotherapy on clinical outcome of CD19 CAR T therapy in adult acute lymphoblastic leukemia. Leukemia 2021, 35, 3268–3271. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, X.; Chen, D.; Yu, J. Radiotherapy combined with immunotherapy: The dawn of cancer treatment. Signal. Transduct. Target Ther. 2022, 7, 258. [Google Scholar] [CrossRef]
- Funk, C.R.; Wang, S.; Chen, K.Z.; Waller, A.; Sharma, A.; Edgar, C.L.; Gupta, V.A.; Chandrakasan, S.; Zoine, J.T.; Fedanov, A.; et al. PI3Kdelta/gamma inhibition promotes human CART cell epigenetic and metabolic reprogramming to enhance antitumor cytotoxicity. Blood 2022, 139, 523–537. [Google Scholar] [CrossRef]
- Zheng, W.; O’Hear, C.E.; Alli, R.; Basham, J.H.; Abdelsamed, H.A.; Palmer, L.E.; Jones, L.L.; Youngblood, B.; Geiger, T.L. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia 2018, 32, 1157–1167. [Google Scholar] [CrossRef]
- Kotla, V.; Goel, S.; Nischal, S.; Heuck, C.; Vivek, K.; Das, B.; Verma, A. Mechanism of action of lenalidomide in hematological malignancies. J. Hematol. Oncol. 2009, 2, 36. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Walter, M.; Urak, R.; Weng, L.; Huynh, C.; Lim, L.; Wong, C.W.; Chang, W.C.; Thomas, S.H.; Sanchez, J.F.; et al. Lenalidomide Enhances the Function of CS1 Chimeric Antigen Receptor-Redirected T Cells Against Multiple Myeloma. Clin. Cancer Res. 2018, 24, 106–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Works, M.; Soni, N.; Hauskins, C.; Sierra, C.; Baturevych, A.; Jones, J.C.; Curtis, W.; Carlson, P.; Johnstone, T.G.; Kugler, D.; et al. Anti-B-cell Maturation Antigen Chimeric Antigen Receptor T cell Function against Multiple Myeloma Is Enhanced in the Presence of Lenalidomide. Mol. Cancer Ther. 2019, 18, 2246–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, E.A.; Alanio, C.; Svoboda, J.; Nasta, S.D.; Landsburg, D.J.; Lacey, S.F.; Ruella, M.; Bhattacharyya, S.; Wherry, E.J.; Schuster, S.J. Pembrolizumab for B-cell lymphomas relapsing after or refractory to CD19-directed CAR T-cell therapy. Blood 2022, 139, 1026–1038. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Lu, W.; Sun, R.; Jin, X.; Cheng, L.; He, X.; Wang, L.; Yuan, T.; Lyu, C.; Zhao, M. Anti-CD19 Chimeric Antigen Receptor T Cells in Combination With Nivolumab Are Safe and Effective Against Relapsed/Refractory B-Cell Non-hodgkin Lymphoma. Front. Oncol. 2019, 9, 767. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Chen, Y.; Jing, Y.; Green, M.R.; Han, L. Advancing CAR T cell therapy through the use of multidimensional omics data. Nat. Rev. Clin. Oncol. 2023, 20, 211–228. [Google Scholar] [CrossRef]
- Dannenfelser, R.; Allen, G.M.; VanderSluis, B.; Koegel, A.K.; Levinson, S.; Stark, S.R.; Yao, V.; Tadych, A.; Troyanskaya, O.G.; Lim, W.A. Discriminatory Power of Combinatorial Antigen Recognition in Cancer T Cell Therapies. Cell Syst. 2020, 11, 215–228. [Google Scholar] [CrossRef]
- Hu, Z.; Yuan, J.; Long, M.; Jiang, J.; Zhang, Y.; Zhang, T.; Xu, M.; Fan, Y.; Tanyi, J.L.; Montone, K.T.; et al. The Cancer Surfaceome Atlas integrates genomic, functional and drug response data to identify actionable targets. Nat. Cancer 2021, 2, 1406–1422. [Google Scholar] [CrossRef]
- Olivier, M.; Asmis, R.; Hawkins, G.A.; Howard, T.D.; Cox, L.A. The Need for Multi-Omics Biomarker Signatures in Precision Medicine. Int. J. Mol. Sci. 2019, 20, 4781. [Google Scholar] [CrossRef] [Green Version]
- Perna, F.; Berman, S.H.; Soni, R.K.; Mansilla-Soto, J.; Eyquem, J.; Hamieh, M.; Hendrickson, R.C.; Brennan, C.W.; Sadelain, M. Integrating Proteomics and Transcriptomics for Systematic Combinatorial Chimeric Antigen Receptor Therapy of AML. Cancer Cell 2017, 32, 506–519. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.H.; Okuma, A.; Sofjan, K.; Lee, S.; Collins, J.J.; Wong, W.W. Engineering advanced logic and distributed computing in human CAR immune cells. Nat. Commun. 2021, 12, 792. [Google Scholar] [CrossRef]
- Garcia-Prieto, C.A.; Villanueva, L.; Bueno-Costa, A.; Davalos, V.; Gonzalez-Navarro, E.A.; Juan, M.; Urbano-Ispizua, A.; Delgado, J.; Ortiz-Maldonado, V.; Del Bufalo, F.; et al. Epigenetic Profiling and Response to CD19 Chimeric Antigen Receptor T-Cell Therapy in B-Cell Malignancies. J. Natl. Cancer Inst. 2022, 114, 436–445. [Google Scholar] [CrossRef]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019, 576, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Park, J.J.; Peng, L.; Yang, Q.; Chow, R.D.; Dong, M.B.; Lam, S.Z.; Guo, J.; Tang, E.; Zhang, Y.; et al. A genome-scale gain-of-function CRISPR screen in CD8 T cells identifies proline metabolism as a means to enhance CAR-T therapy. Cell Metab. 2022, 34, 595–614. [Google Scholar] [CrossRef]
- Blache, U.; Weiss, R.; Boldt, A.; Kapinsky, M.; Blaudszun, A.R.; Quaiser, A.; Pohl, A.; Miloud, T.; Burgaud, M.; Vucinic, V.; et al. Advanced Flow Cytometry Assays for Immune Monitoring of CAR-T Cell Applications. Front. Immunol. 2021, 12, 658314. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, L.; Haas, E.R.; Vyas, V.; Urak, R.; Forman, S.J.; Wang, X. Single-cell analysis by mass cytometry reveals CD19 CAR T cell spatiotemporal plasticity in patients. OncoImmunology 2022, 11, 2040772. [Google Scholar] [CrossRef] [PubMed]
- Volpe, A.; Nagle, V.L.; Lewis, J.S.; Ponomarev, V. Predicting CAR-T cell Immunotherapy Success through ImmunoPET. Clin. Cancer Res. 2021, 27, 911–912. [Google Scholar] [CrossRef]
- Simonetta, F.; Alam, I.S.; Lohmeyer, J.K.; Sahaf, B.; Good, Z.; Chen, W.; Xiao, Z.; Hirai, T.; Scheller, L.; Engels, P.; et al. Molecular Imaging of Chimeric Antigen Receptor T Cells by ICOS-ImmunoPET. Clin. Cancer Res. 2021, 27, 1058–1068. [Google Scholar] [CrossRef]
Figure 1.
Endogenous T cell dysfunction and cancer-cell-intrinsic alterations contribute to cancer progression and failure of immunotherapy. A dynamic balance exists between tumor cells and T cells—T cells work to prevent tumors from growing and spreading, while tumor cells proliferate and metastasize, attempting to surmount immune attack. Cancer progresses when this homeostasis is broken, and tumor cell growth prevails. This figure illustrates the two distinct sets of factors that play a role in cancer progression—endogenous T cell dysfunction, illustrated by the eight panels on the right side of the figure, and cancer-cell-intrinsic alterations, depicted by the eight panels on the left side of the figure.
Figure 1.
Endogenous T cell dysfunction and cancer-cell-intrinsic alterations contribute to cancer progression and failure of immunotherapy. A dynamic balance exists between tumor cells and T cells—T cells work to prevent tumors from growing and spreading, while tumor cells proliferate and metastasize, attempting to surmount immune attack. Cancer progresses when this homeostasis is broken, and tumor cell growth prevails. This figure illustrates the two distinct sets of factors that play a role in cancer progression—endogenous T cell dysfunction, illustrated by the eight panels on the right side of the figure, and cancer-cell-intrinsic alterations, depicted by the eight panels on the left side of the figure.

Figure 2.
The dual contribution of tumor-associated cells to both cancer progression and T cell dysfunction. Numerous factors present in the TME play a role in driving cancer progression. Tumor-associated cells, such as TAMs, TANs, and CAFs, secrete cytokines and other factors, including TGF-β, IL-6, and ARG-1, which not only inhibit T cell function but also act on cancer cells to promote tumor growth, metastasis, angiogenesis, EMT, and ECM remodeling. Additionally, physical characteristics of the TME, such as depleted nutrients, oxygen, and essential ions, as well as elevated levels of metabolites such as ROS, cholesterol, and lactic acid, further contribute to T cell dysfunction while favoring tumor growth.
Figure 2.
The dual contribution of tumor-associated cells to both cancer progression and T cell dysfunction. Numerous factors present in the TME play a role in driving cancer progression. Tumor-associated cells, such as TAMs, TANs, and CAFs, secrete cytokines and other factors, including TGF-β, IL-6, and ARG-1, which not only inhibit T cell function but also act on cancer cells to promote tumor growth, metastasis, angiogenesis, EMT, and ECM remodeling. Additionally, physical characteristics of the TME, such as depleted nutrients, oxygen, and essential ions, as well as elevated levels of metabolites such as ROS, cholesterol, and lactic acid, further contribute to T cell dysfunction while favoring tumor growth.

Figure 3.
The landscape of cancer therapeutics. This figure illustrates the various therapeutics that are currently in use for the treatment of cancer. These include traditional therapies such as surgery, chemotherapy, and radiotherapy, targeted therapies including small molecule drugs, monoclonal antibodies and derivatives, oncolytic viruses, and gene editing, as well as immunotherapies including cancer vaccines, checkpoint inhibitors, and engineered T cell therapies.
Figure 3.
The landscape of cancer therapeutics. This figure illustrates the various therapeutics that are currently in use for the treatment of cancer. These include traditional therapies such as surgery, chemotherapy, and radiotherapy, targeted therapies including small molecule drugs, monoclonal antibodies and derivatives, oncolytic viruses, and gene editing, as well as immunotherapies including cancer vaccines, checkpoint inhibitors, and engineered T cell therapies.


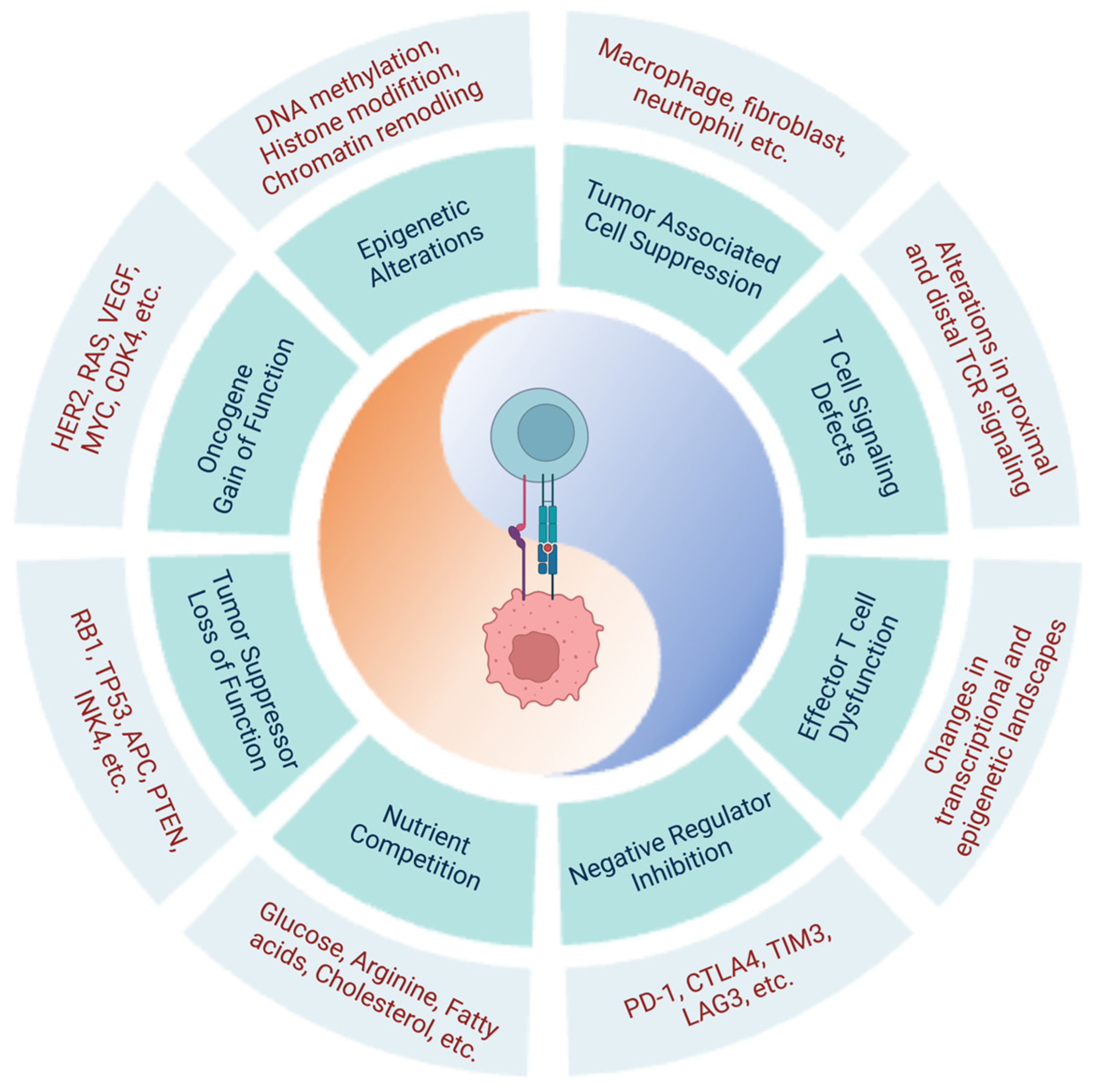{kind=link}
{kind=link}
{kind=link}
Table 1.
Current ongoing or completed clinical trials of CAR T and TCR T therapies *.
| NCT ID | Official Study Title | Target Antigen | Trial Phase | Targeted Conditions | Related Publications |
|---|---|---|---|---|---|
| Clinical trials of CAR T therapies | |||||
| NCT04030195 | A Phase 1/2a, Open-label, Dose-escalation, Dose-expansion, Parallel Assignment Study to Evaluate the Safety and Clinical Activity of PBCAR20A in Subjects with Relapsed/Refractory (r/r) Non-Hodgkin Lymphoma (NHL), or r/r Chronic Lymphocytic Leukemia (CLL), or Small Lymphocytic Lymphoma (SLL) | Cluster of differentiation 20 (CD20) | Phase I/IIa | Non-Hodgkin lymphoma, chronic lymphocytic leukemia, small lymphocytic lymphoma | |
| NCT03958656 | A Phase I Clinical Trial of T-cells Expressing an Anti-SLAMF7 CAR for Treating Multiple Myeloma | Signaling lymphocytic activation molecule F7 (SLAM7) | Phase I | Multiple myeloma | |
| NCT03338972 | A Phase I Study of Adoptive Immunotherapy for Advanced B Cell Maturation Antigen (BCMA)+ Multiple Myeloma with Autologous CD4+ and CD8+ T Cells Engineered to Express a BCMA-Specific Chimeric Antigen Receptor | BCMA | Phase I | Multiple myeloma | |
| NCT03289455 | A Single-Arm, Open-Label, Multi-Centre, Phase I/II Study Evaluating the Safety and Clinical Activity Of AUTO3, a CAR T Cell Treatment Targeting CD19 And CD22 in Pediatric And Young Adult Patients With Relapsed or Refractory B Cell Acute Lymphoblastic Leukemia | CD19, CD22 | Phase I/II | B cell acute lymphoblastic leukemias | |
| NCT03049449 | Anti-CD30 CAR T Cells with Fully-human Binding Domains for Treating CD30-expressing Lymphomas Including Anaplastic Large Cell Lymphomas | CD30 | Phase 1 | Lymphomas | |
| NCT03019055 | Phase 1/1b Study of Redirected Autologous T Cells Engineered to Contain an Anti CD19 and Anti CD20 scFv Coupled to CD3ζ and 4-1BB Signaling Domains in Patients with Relapsed and/or Refractory CD19 or CD20 Positive B Cell Malignancies | CD19, CD20 | Phase I/Ib | Lymphoma, non-Hodgkin lymphoma, B cell chronic lymphocytic leukemia, small lymphocytic lymphoma | |
| NCT02761915 | A Cancer Research UK Phase I Trial of Anti-GD2 Chimeric Antigen Receptor (CAR) Transduced T-cells (1RG-CART) in Patients with Relapsed or Refractory Neuroblastoma | disialoganglioside (GD2) | Phase I | Neuroblastoma | [14] |
| NCT02659943 | T Cells Expressing a Fully-Human Anti-CD19 Chimeric Antigen Receptor for Treating B cell Malignancies | CD19 | Phase I | B cell lymphoma, non-Hodgkin’s lymphoma | [15] |
| NCT02215967 | A Phase I Clinical Trial of T-Cells Targeting B Cell Maturation Antigen for Previously Treated Multiple Myeloma | BCMA | Phase I | Multiple myeloma | [16,17,18] |
| NCT02030847 | Phase II Study of Redirected Autologous T Cells Engineered to Contain Anti-CD19 Attached to TCR and 4-1BB Signaling Domains in Patients with Chemotherapy Resistant or Refractory Acute Lymphoblastic Leukemia | CD19 | Phase II | B cell acute lymphoblastic leukemias | [19] |
| NCT01583686 | Phase I/II Study of Metastatic Cancer Using Lymphodepleting Conditioning Followed by Infusion of Anti-mesothelin Gene Engineered Lymphocytes | Mesothelin | Phase I/II | Cervical, pancreatic, ovarian, lung cancers; mesothelioma | [20,21] |
| NCT00924287 | Phase I/II Study of Metastatic Cancer That Expresses Her-2 Using Lymphodepleting Conditioning Followed by Infusion of Anti-Her-2 Gene Engineered Lymphocytes | HER-2 | Phase I/II | Metastatic cancers | [22,23] |
| NCT01460901 | Phase I Study of Donor Derived, Gene Modified, Multi-virus-specific, Cytotoxic T-Lymphocytes Redirected to GD2 for Relapsed/Refractory Neuroblastoma Post-allo Stem Cell Transplantation with Submyeloblative Conditioning | disialoganglioside (GD2) | Phase I | Neuroblastoma | |
| NCT01454596 | A Phase I/II Study of the Safety and Feasibility of Administering T Cells Expressing Anti-EGFRvIII Chimeric Antigen Receptor to Patients with Malignant Gliomas Expressing EGFRvIII | Epidermal growth factor receptor variant III (EGFRvIII) | Phase I/II | Malignant glioma, glioblastoma, brain cancer, gliosarcoma | |
| NCT01029366 | Pilot Study of Redirected Autologous T-cells Engineered to Contain Anti-CD19 Attached to TCR and 4-1BB Signaling Domains in Patient with Chemotherapy Resistant or Refractory CD19+ Leukemia and Lymphoma | CD19 | Phase I | Leukemias, lymphomas | [19,24,25] |
| Clinical trials of TCR T therapies | |||||
| NCT02992743 | A Pilot Study of NY-ESO-1c259T Cells in Subjects with Advanced Myxoid/Round Cell Liposarcoma | New York esophageal antigen-1 (NY-ESO-1) | Phase II | Liposarcoma | |
| NCT02650986 | A Phase I/IIa Study of TGFß Blockade in TCR-Engineered T Cell Cancer Immunotherapy in Patients with Advanced Malignancies | NY-ESO-1 | Phase I/IIa | Various carcinomas, neoplasms, sarcomas | |
| NCT02588612 | A Pilot Open-Label Clinical Trial Evaluating the Safety and Efficacy of Autologous T Cells Expressing Enhanced TCRs Specific for NY-ESO-1 in Subjects with Stage IIIb or Stage IV Non-Small Cell Lung Cancer (NSCLC) | NY-ESO-1 | Phase I | Non-small cell lung cancer | |
| NCT02280811 | A Phase I/II Study of T Cell Receptor Gene Therapy Targeting HPV-16 E6 for HPV-Associated Cancers | HPV-16 E6 | Phase I/II | (HPV)-16+ cancers (cervical, vulvar, vaginal, penile, anal, and oropharyngeal cancers) | [26,27,28] |
| NCT02111850 | A Phase I/II Study of the Treatment of Metastatic Cancer That Expresses MAGE-A3 Using Lymphodepleting Conditioning Followed by Infusion of HLA-DP0401/0402 Restricted Anti-MAGE-A3 TCR-Gene Engineered Lymphocytes and Aldesleukin | Melanoma antigen family A, 3 (MAGE-A3) | Phase I/II | Cervical, renal, urothelial, breast cancers; melanoma | |
| NCT04015336 | A Phase II Study of E7 TCR T Cell Induction Immunotherapy for Stage II and Stage III HPV-Associated Oropharyngeal Cancer | E7 (HPV oncoprotein) | Phase II | Papillomavirus infections, oropharyngeal neoplasms | |
| NCT01567891 | A Phase I/IIa, Open Label Clinical Trial Evaluating the Safety and Efficacy of Autologous T Cells Expressing Enhanced TCRs Specific for NY-ESO-1 in Patients with Recurrent or Treatment Refractory Ovarian Cancer. | NY-ESO-1 | Phase I/IIa | Ovarian cancer | |
| NCT00393029 | Phase II Study of Metastatic Cancer That Overexpresses p53 Using Lymphodepleting Conditioning Followed by Infusion of Anti-p53 T Cell Receptor (TCR)-Gene Engineered Lymphocytes | Protein 53 (p53) | Phase II | Metastatic cancers overexpressing p53 | |
| NCT01343043 | A Pilot Study of Genetically Engineered NY-ESO-1 Specific NY-ESO-1ᶜ2⁵⁹T in HLA-A2+ Patients with Synovial Sarcoma (NY-ESO-1) | NY-ESO-1 | Phase I | Neoplasms | [29,30,31] |
| NCT00509288 | Phase II Study of Metastatic Melanoma Using Lymphodepleting Conditioning Followed by Infusion of Anti-MART-1 F5 TCR-Gene Engineered Lymphocytes | Melanoma antigen recognized by T-cells (MART-1) | Phase II | Melanoma, skin cancer | [32,33,34,35] |
* Updated April 2023.
Table 2.
Alterations in negative regulator expression and changes in the transcriptional and epigenetic landscape of T cells contribute to immune dysfunction.
Table 2.
Alterations in negative regulator expression and changes in the transcriptional and epigenetic landscape of T cells contribute to immune dysfunction.
| Molecule | Alteration | Consequence | Associated Malignancy | References | ||
|---|---|---|---|---|---|---|
| Negative regulators | Cell Surface | PD-1 | Overexpressed in T cells upon chronic antigen exposure; denotes exhausted phenotype | Limited T cell survival and function; associated with T cell dysfunction | Oncogenesis | [11,12,48,51,56,57,58,63,81,88,89,90,91,92,93,94,95,96,97,99,128,130,146] |
| TIM-3 | ||||||
| LAG-3 | ||||||
| CTLA-4 | ||||||
| BTLA | ||||||
| TIGIT | ||||||
| CD244 | ||||||
| CD160 | ||||||
| Intracellular | SHP-1 | Decreased expression of SHP-1 | Hyperactive TYK2 and JAK1 kinases | Lymphomagenesis | [106,107] | |
| SHP-2 | Lower levels of phosphorylated SHP-2 | Metastasis; increased release of inflammatory cytokines; accumulation of MDSCs | Melanoma (murine model) | [108] | ||
| Transcription & epigenetic factors | DUSP2 (PAC1) | Highly expressed in TILs | Recruitment of Mi-2β nucleosome-remodeling and histone-deacetylase complex; facilitated T cell exhaustion and loss of proliferative and effector functions | Various cancers | [134,135] | |
| Blimp-1 | Overexpressed in exhausted CD8+ T cells | Increased expression of inhibitory receptors; repression of memory CD8+ T cell differentiation | Various cancers | [54,84,128,131,132,146,147] | ||
| EOMES | Directly controls expression of TIM-3; antagonization of T-bet | |||||
| BATF | Expression of inhibitory receptors; repression of TCF1, a transcription factor required for memory T cell differentiation | |||||
| T-bet | Decreased expression in dysfunctional T cells | T-bet directly represses expression of PD1 | Various cancers | [54,128,129] | ||
| TOX | Critical for CD8+ T cell exhaustion | Required for gene expression of inhibitory receptors (e.g., Pdcd1, Entpd1, Havcr2, Cd244, and Tigit) | Various cancers | [54,55,57,98,118,119,120,121] | ||
| NR4A1 | Expressed at high levels in tolerant T cells | Various cancers | [110,112,113,114,115,116] | |||
| Critical for development of dysfunctional exhausted T cells | Attenuated T cell effector functions and upregulation of inhibitory receptors | [54,55,57,98,118,119,120,121] | ||||
| TET2 | Mutations | Heightened hypermethylation of T cell signaling/differentiation genes | Lymphomas | [75,77,78,143,144,145] | ||
| DNMT3A | ||||||
| IDH2 |
Table 3.
Key molecular mechanisms of oncogenesis in tumor cells.
| Gene | Tumor Type | Reference | ||
|---|---|---|---|---|
| Genetics | Oncogenes | BCL2 | B cell leukemia, lymphoma | [148] |
| BCR/ABL | Leukemia | [149,150] | ||
| BRAF | Melanoma, papillary thyroid carcinoma | [151] | ||
| CDK4 | Various tumors | [152] | ||
| C-KIT | Acute myeloid leukemia, myelodysplastic syndrome (MDS), MDS-derived AML | [153] | ||
| EGFR | Lung tumor | [154,155] | ||
| ERBA | Breast cancer | [156] | ||
| FGF5 | Hepatocellular carcinoma, colorectal cancer, prostate cancer | [157,158] | ||
| FMS | Feline McDonough sarcoma | [159] | ||
| FOS | HNSCC, breast cancer, various tumors | [160] | ||
| GLI | Various tumors | [161] | ||
| HER2 | Breast tumor | [162] | ||
| HST | Esophageal cancer, stomach cancer | [163] | ||
| JUN | Various tumors | [164] | ||
| MDM2 | Various tumors | [165] | ||
| MET | Hereditary renal papillary carcinoma | [166] | ||
| MYC | Various tumors | [167] | ||
| RAS | Various tumors | [168,169,170,171] | ||
| RET | Papillary thyroid carcinoma | [172] | ||
| SIS | Breast tumor | [173] | ||
| SOS | Ras-related cancer | [174] | ||
| SRC | Various tumors | [175] | ||
| TTG | Various tumors | [176] | ||
| VEGF | Angiogenesis | [177] | ||
| VEGFR | Migration of cancer cells | [178,179] | ||
| Tumor suppressors | APC | Colon/rectum carcinoma, adenomatous polyposis | [180,181] | |
| ARF (p14) | Melanoma | [181] | ||
| BRCA1, BRCA2 | Breast, ovarian, pancreatic carcinomas | [180,181,182,183] | ||
| CDH1 | Gastric cancer | [181] | ||
| CHK 1/2 | Li-Fraumeni syndrome | [181] | ||
| DCC | Colorectal carcinoma | [181] | ||
| DPC4 | Pancreatic carcinoma | [180] | ||
| INK4 (p16) | Melanoma, lung carcinoma, brain tumors, leukemia, lymphoma | [180,181] | ||
| MADR2 | Colon/rectum carcinoma | [180] | ||
| MLH1, MSH2, MSH6 | Colorectal cancer | [181] | ||
| NF1 | Neurofibrosarcoma, neurofibromatosis type I | [180,181] | ||
| NF2 | Meningioma | [180] | ||
| PTC | Basal cell carcinoma | [180] | ||
| PTEN | Brain tumors; melanoma; prostate, endometrial, kidney, lung carcinomas | [180] | ||
| RB1 | Retinoblastoma | [184] | ||
| TP53 | Brain tumors; breast, colon/rectum, esophageal, liver, lung carcinomas; sarcomas; leukemias, lymphomas, Li-Fraumeni syndrome | [180,181] | ||
| VHL | Renal cell carcinoma | [180] | ||
| WT1, WT2 | Wilms’ tumor | [180,185] | ||
| Epigenetics | DNA methylation | AID | Chronic myeloid leukemia | [186] |
| DNMT1 | Colorectal, non-small-cell lung, pancreatic, gastric, breast cancer | [187,188] | ||
| DNMT3A | Myelodysplastic syndromes, acute myeloid leukemia | [189,190,191] | ||
| DNMT3B | ICF syndrome, SNPs in breast and lung adenoma | [192,193] | ||
| IDH1/2 | Glioma, acute myeloid leukemia | [194,195,196] | ||
| MADR2 | Colon/rectum carcinoma | [180] | ||
| MBD1/2 | Lung cancer, breast cancer | [197] | ||
| MLH1, MSH2, MSH6 | Colorectal cancer | [181] | ||
| MLL1/2/3 | Bladder TCC, hematopoietic, non-Hodgkin lymphoma, B cell lymphoma, prostate cancer | [198,199] | ||
| Histone modifiers | BMI-1 | Ovarian, mantle cell lymphomas, Merkel cell carcinomas | [200,201] | |
| CREBBP (CBP/KAT3A) | Gastric and colorectal, epithelial, ovarian, lung, esophageal cancer | [202] | ||
| EP300 (P300/KAT3B) | Breast, colorectal, pancreatic cancer | [202] | ||
| EZH2 | Breast, prostate, bladder, colon, pancreas, liver, gastric, uterine tumors, melanoma, lymphoma, myeloma, Ewing’s sarcoma | [203,204] | ||
| G9a | HCC, cervical, uterine, ovarian, breast cancer | [205] | ||
| HDAC2 | Colonic, gastric, endometrial cancer | [206] | ||
| JARID1B/C (KDM5C) | Testicular cancer, breast cancer, RCCC | [207] | ||
| LSD1 | Prostate | [207] | ||
| PCAF | Epithelial | [202] | ||
| PRMT1/5 | Breast, gastric cancer | [202] | ||
| SIRT1, HDAC5/7A | Breast, colorectal, prostate cancer | [202] | ||
| UTX (KDM6A) | Bladder, breast, kidney, lung, pancreas, esophagus, colon, uterus, brain, hematological malignancies | [207] | ||
| Chromatin remodelers | ARID1A (BAF250A) | Ovarian clear cell carcinomas, endometrioid carcinomas, endometrial carcinomas | [208,209] | |
| ARID2 (BAF200) | Primary pancreatic adenocarcinomas | [210] | ||
| BRD7 | Bladder TCC | [211] | ||
| BRM (SMARCA2) | Prostate, basal cell carcinoma | [212,213] | ||
| CHD4/5 | Colorectal and gastric cancer, ovarian, prostate, neuroblastoma, hematopoietic | [214,215,216] | ||
| CHD7 | Gastric and colorectal cancer | [217] | ||
| P400/Tip60 | Lymphoma, colon, head and neck, breast cancer | [218] | ||
| PBRM1 (BAF180) | Breast tumors | [219] | ||
| SNF5 (SMARCB1, INI1) | Lung, rhabdoid, medulloblastoma | [220] | ||
| SRCAP | Prostate | [221] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chen, C.; Liu, X.; Chang, C.-Y.; Wang, H.Y.; Wang, R.-F. The Interplay between T Cells and Cancer: The Basis of Immunotherapy. Genes 2023, 14, 1008. https://doi.org/10.3390/genes14051008
AMA Style
Chen C, Liu X, Chang C-Y, Wang HY, Wang R-F. The Interplay between T Cells and Cancer: The Basis of Immunotherapy. Genes. 2023; 14(5):1008. https://doi.org/10.3390/genes14051008
Chicago/Turabian StyleChen, Christina, Xin Liu, Che-Yu Chang, Helen Y. Wang, and Rong-Fu Wang. 2023. "The Interplay between T Cells and Cancer: The Basis of Immunotherapy" Genes 14, no. 5: 1008. https://doi.org/10.3390/genes14051008
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.