

Evolutionary Rates, Divergence Rates, and Performance of Individual Mitochondrial Genes Based on Phylogenetic Analysis of Copepoda

Abstract
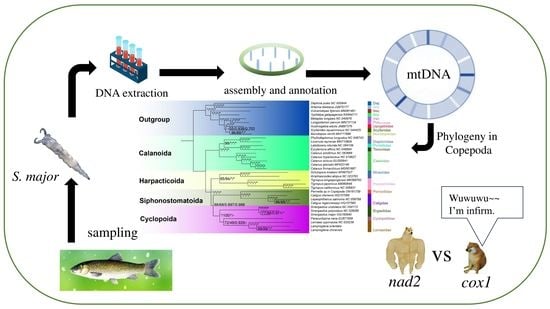
1. Introduction
2. Materials and Methods
2.1. Sampling and Identification
2.2. Mitochondrial Genome Sequencing and Assembly
2.3. Genome Composition and Sequence Analyses
2.4. Phylogenetic Analyses
2.5. Phylogenetic Examination of Separate Genes
3. Results
3.1. The Structure of the Mitochondrial Genome
3.2. Comparative Analysis of Copepoda Mitogenomes
3.3. Nucleotide Diversity and Evolutionary Rate Analysis
3.4. Phylogenetic Analyses
3.5. Phylogenetic Examination of Individual Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar]
- Yuan, B.; He, G.; Dong, W. The first complete mitochondrial genome of the genus Echinolaelaps reveals mitochondrial genome rearrangement type and evolution of Gamasida. Parasitology 2023, 150, 644–652. [Google Scholar] [CrossRef]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L.; Lambkin, C.L.; Barker, S.C.; Whiting, M.F. A mitochondrial genome phylogeny of Diptera: Whole genome sequence data accurately resolve relationships over broad timescales with high precision. Syst. Entomol. 2007, 32, 40–59. [Google Scholar] [CrossRef]
- Li, H.; Leavengood, J.M., Jr.; Chapman, E.G.; Burkhardt, D.; Song, F.; Jiang, P.; Liu, J.; Zhou, X.; Cai, W. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc. R. Soc. B Biol. Sci. 2017, 284, 20171223. [Google Scholar] [CrossRef]
- Du, Z.; Hasegawa, H.; Cooley, J.R.; Simon, C.; Yoshimura, J.; Cai, W.; Sota, T.; Li, H. Mitochondrial genomics reveals shared phylogeographic patterns and demographic history among three periodical cicada species groups. Mol. Biol. Evol. 2019, 36, 1187–1200. [Google Scholar] [CrossRef]
- Mueller, R.L. Evolutionary rates, divergence dates, and the performance of mitochondrial genes in Bayesian phylogenetic analysis. Syst. Biol. 2006, 55, 289–300. [Google Scholar] [CrossRef]
- Aladetohun, N.F.; Sakiti, N.G.; Babatunde, E.E. Copepoda parasites in economically important fish, Mugilidae (Mugil cephalus and Liza falcipinnis from Lac Nokoue Lagoon in Republic of Benin, West Africa. Afr. J. Environ. Sci. Technol. 2013, 7, 799–807. [Google Scholar]
- Boxshall, G.; Defaye, D. Global diversity of copepods (Crustacea: Copepoda) in freshwater. Hydrobiologia 2007, 595, 195–207. [Google Scholar] [CrossRef]
- Cakic, P.; Lenhardt, M.; Kolarevic, J. Sinergasilus polycolpus, a new copepod species in the ichthyoparasitofauna of Serbia and Montenegro. Dis. Aquat. Org. 2004, 58, 265–266. [Google Scholar] [CrossRef]
- Molnár, K.; Székely, C. Occurrence and pathology of Sinergasilus lieni (Copepoda: Ergasilidae), a parasite of the silver carp and bighead, in Hungarian ponds. Acta Vet. Hung. 2004, 52, 51–60. [Google Scholar] [CrossRef]
- Blazhekovikj-Dimovska, D.; Stojanovski, S. Grass Carp (Ctenopharyngodon idella Valenciennes, 1844)(Pisces: Cyprinidae) as host of new parasite species in the Ichthyoparasitofauna of Macedonia. J. BioScience Biotechnol. 2020, 9, 29–35. [Google Scholar]
- Yevtushenko, A. Features of the parasitic system formation in herbivorous fis h in the aquaculture of the North-Eastern and Eastern regions of Ukraine. J. Vet. Med. Biotechnol. Biosaf. 2020, 6, 18–24. [Google Scholar] [CrossRef]
- Song, Y. Phylogenetic Study of Parasitic Copepods in the Ergasilidae and Lernaeidae on Freshwater Fish in China; Institute of Hydrobiology, Chinese Academy of Sciences: Wuhan, China, 2006. [Google Scholar]
- Mandygra, M. Mixed crustaceosis infestation at black-head minnows. Bull. Agric. Sci. 2017, 95. [Google Scholar] [CrossRef]
- Soares, I.A.; Salinas, V.; Ponti, O.d.; Mancini, M.A.; Luque, J.L. First molecular data for Lernaea cyprinacea (Copepoda: Cyclopoida) infesting Odontesthes bonariensis, a commercially important freshwater fish in Argentina. Rev. Bras. Parasitol. Vet. 2018, 27, 105–108. [Google Scholar] [CrossRef]
- Stock, J. Parasitic Copepoda of British Fishes; JSTOR: New York, NY, USA, 1981. [Google Scholar]
- Khodami, S.; Mercado-Salas, N.F.; Tang, D.; Arbizu, P.M. Molecular evidence for the retention of the Thaumatopsyllidae in the order Cyclopoida (Copepoda) and establishment of four suborders and two families within the Cyclopoida. Mol. Phylogen Evol. 2019, 138, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, F.D.; von Vaupel Klein, J.C. Rhabdomoplea, a new superorder for the thaumatopsylloid copepods: The consequence of an alternative hypothesis of copepod phylogeny. Crustaceana 2019, 92, 177–188. [Google Scholar] [CrossRef]
- Kern, E.M.; Kim, T.; Park, J.-K. The mitochondrial genome in nematode phylogenetics. Front. Ecol. Evol. 2020, 8, 250. [Google Scholar] [CrossRef]
- Skern-Mauritzen, R.; Torrissen, O.; Glover, K.A. Pacific and Atlantic Lepeophtheirus salmonis (Krøyer, 1838) are allopatric subspecies: Lepeophtheirus salmonis salmonis and L. salmonis oncorhynchi subspecies novo. BMC Genet. 2014, 15, 32. [Google Scholar] [CrossRef]
- Parshukov, A.; Vlasenko, P.; Simonov, E.; Ieshko, E.; Burdukovskaya, T.; Anikieva, L.; Kashinskaya, E.; Andree, K.B.; Solovyev, M. Parasitic copepods Caligus lacustris (Copepoda: Caligidae) on the rainbow trout Oncorhynchus mykiss in cage aquaculture: Morphology, population demography, and first insights into phylogenetic relationships. Parasitol. Res. 2021, 120, 2455–2467. [Google Scholar] [CrossRef]
- Zaidykov, I.; Bukin, Y.; Naumova, E.; Kirilchik, S.; Sukhanova, L. Phylogenetic relationships and historical population reconstruction of Asian members of the genus Epischura (Copepoda, Calanoida). J. Great Lakes Res. 2020, 46, 12–16. [Google Scholar] [CrossRef]
- Easton, E.E.; Darrow, E.M.; Spears, T.; Thistle, D. The mitochondrial genomes of Amphiascoides atopus and Schizopera knabeni (Harpacticoida: Miraciidae) reveal similarities between the copepod orders Harpacticoida and Poecilostomatoida. Gene 2014, 538, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Havird, J.C.; Santos, S.R. Performance of single and concatenated sets of mitochondrial genes at inferring metazoan relationships relative to full mitogenome data. PLoS ONE 2014, 9, e84080. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, H.; Song, F.; Zhao, Y.; Wilson, J.J.; Cai, W. Higher-level phylogeny and evolutionary history of Pentatomomorpha (Hemiptera: Heteroptera) inferred from mitochondrial genome sequences. Syst. Entomol. 2019, 44, 810–819. [Google Scholar] [CrossRef]
- Nadimi, M.; Daubois, L.; Hijri, M. Mitochondrial comparative genomics and phylogenetic signal assessment of mtDNA among arbuscular mycorrhizal fungi. Mol. Phylogen Evol. 2016, 98, 74–83. [Google Scholar] [CrossRef]
- Kuang, P.; Qian, J. Economic Fauna of China: Parasitic Crustacea of Freshwater Fishes; Science Press: Beijing, China, 1991. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogen Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovli, I.; Zou, H.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Garrett, R.C.; Nar, A.; Fisher, T.J.; Maurer, K. ggvoronoi: Voronoi diagrams and heatmaps with ggplot2. J. Open Source Softw. 2018, 3, 1096. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Lemmon, A.R.; Brown, J.M.; Stanger-Hall, K.; Lemmon, E.M. The effect of ambiguous data on phylogenetic estimates obtained by maximum likelihood and Bayesian inference. Syst. Biol. 2009, 58, 130–145. [Google Scholar] [CrossRef]
- Heath, T.A.; Hedtke, S.M.; Hillis, D.M. Taxon sampling and the accuracy of phylogenetic analyses. J. Syst. Evol. 2008, 46, 239–257. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Lanfear, R.; Calcott, B.; Ho, S.Y.; Guindon, S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]
- Soria-Carrasco, V.; Talavera, G.; Igea, J.; Castresana, J. The K tree score: Quantification of differences in the relative branch length and topology of phylogenetic trees. Bioinformatics 2007, 23, 2954–2956. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Moritz, C.; Brown, W.M. Tandem duplications in animal mitochondrial DNAs: Variation in incidence and gene content among lizards. Proc. Natl. Acad. Sci. USA 1987, 84, 7183–7187. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Brown, W.M.; Boore, J.L. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc. Natl. Acad. Sci. USA 2000, 97, 13738–13742. [Google Scholar] [CrossRef]
- Huys, R.; Llewellyn-Hughes, J.; Olson, P.D.; Nagasawa, K. Small subunit rDNA and Bayesian inference reveal Pectenophilus ornatus (Copepoda incertae sedis) as highly transformed Mytilicolidae, and support assignment of Chondracanthidae and Xarifiidae to Lichomolgoidea (Cyclopoida). Biol. J. Linn. Soc. 2006, 87, 403–425. [Google Scholar] [CrossRef]
- Hua, C.J.; Su, M.Y.; Sun, Z.W.; Lu, Y.H.; Feng, J.M. Complete mitochondrial genome of the copepod Sinergasilus undulates (Copepoda: Poecilostomatoida). Mitochondrial DNA Part B 2021, 6, 1226–1228. [Google Scholar] [CrossRef]
- Minxiao, W.; Song, S.; Chaolun, L.; Xin, S. Distinctive mitochondrial genome of Calanoid copepod Calanus sinicus with multiple large non-coding regions and reshuffled gene order: Useful molecular markers for phylogenetic and population studies. BMC Genom. 2011, 12, 73. [Google Scholar] [CrossRef]
- Austin, C.M.; Tan, M.H.; Lee, Y.P.; Croft, L.J.; Meekan, M.G.; Pierce, S.J.; Gan, H.M. The complete mitogenome of the whale shark parasitic copepod Pandarus rhincodonicus norman, Newbound & Knott (Crustacea; Siphonostomatoida; Pandaridae)—A new gene order for the copepoda. Mitochondrial DNA 2014, 27, 694–695. [Google Scholar]
- Jakovlić, I.; Zou, H.; Zhao, X.-M.; Zhang, J.; Wang, G.-T.; Zhang, D. Evolutionary history of inversions in directional mutational pressures in crustacean mitochondrial genomes: Implications for evolutionary studies. Mol. Phylogen Evol. 2021, 164, 107288. [Google Scholar] [CrossRef] [PubMed]
- Naylor, G.J.; Collins, T.M.; Brown, W.M. Hydrophobicity and phylogeny. Nature 1995, 373, 565–566. [Google Scholar] [CrossRef]
- Ros, V.I.; Breeuwer, J.A. Spider mite (Acari: Tetranychidae) mitochondrial COI phylogeny reviewed: Host plant relationships, phylogeography, reproductive parasites and barcoding. Exp. Appl. Acarol. 2007, 42, 239–262. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.O.; DeWaard, J.R.; Boutillier, J.; Ratnasingham, S.; Dooh, R.T.; Hajibabaei, M.; Hebert, P.D. Biological identifications through DNA barcodes: The case of the Crustacea. Can. J. Fish. Aquat. Sci. 2007, 64, 272–295. [Google Scholar] [CrossRef]
- Goldstein, D.B.; Ruiz Linares, A.; Cavalli-Sforza, L.L.; Feldman, M.W. An evaluation of genetic distances for use with microsatellite loci. Genetics 1995, 139, 463–471. [Google Scholar] [CrossRef]
- Ye, F.; Liu, T.; Zhu, W.; You, P. Complete mitochondrial genome of Whitmania laevis (Annelida, Hirudinea) and comparative analyses within Whitmania mitochondrial genomes. Belg. J. Zool. 2015, 145, 114–128. [Google Scholar] [CrossRef]
- Ho, J.S. Copepod phylogeny: A reconsideration of Huys & Boxshall’s ‘parsimony versus homology’. In Ecology and Morphology of Copepods; Springer: Berlin/Heidelberg, Germany, 1994; pp. 31–39. [Google Scholar]
- Huys, R.; Boxshall, G.A. Copepod Evolution; The Ray Society: London, UK, 1991. [Google Scholar]
- Mikhailov, K.V.; Ivanenko, V.N. Low support values and lack of reproducibility of molecular phylogenetic analysis of Copepoda orders. Arthropoda Sel. 2021, 30, 39–42. [Google Scholar] [CrossRef]
- Eyun, S.I. Phylogenomic analysis of Copepoda (Arthropoda, Crustacea) reveals unexpected similarities with earlier proposed morphological phylogenies. BMC Evol. Biol. 2017, 17, 23. [Google Scholar] [CrossRef]
- Dahms, H.-U. Postembryonic apomorphies proving the monophyletic status of the Copepoda. Zool. Stud. 2004, 43, 446–453. [Google Scholar]
- Regier, J.C.; Shultz, J.W.; Zwick, A.; Hussey, A.; Ball, B.; Wetzer, R.; Martin, J.W.; Cunningham, C.W. Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature 2010, 463, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Oakley, T.H.; Wolfe, J.M.; Lindgren, A.R.; Zaharoff, A.K. Phylotranscriptomics to bring the understudied into the fold: Monophyletic Ostracoda, fossil placement, and pancrustacean phylogeny. Mol. Biol. Evol. 2013, 30, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, T.S.; Petersen, B.; Petersen, H.C.B.; Browne, P.D.; Prost, S.; Stillman, J.H.; Hansen, L.H.; Hansen, B.W. The genome and mRNA transcriptome of the cosmopolitan calanoid copepod Acartia tonsa Dana improve the understanding of copepod genome size evolution. Genome Biol. Evol. 2019, 11, 1440–1450. [Google Scholar] [CrossRef]
- Zhang, D.; Yan, L.; Zhang, M.; Chu, H.; Cao, J.; Li, K.; Hu, D.; Pape, T. Phylogenetic inference of calyptrates, with the first mitogenomes for Gasterophilinae (Diptera: Oestridae) and Paramacronychiinae (Diptera: Sarcophagidae). Int. J. Biol. Sci. 2016, 12, 489. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Class | Order | Family | ID | ||
|---|---|---|---|---|---|---|
| subgroup_1 | subgroup_2 | outgroups | ||||
| Vulcanolepas fijiensis (Chan, Ju & Kim, 2019) | Thecostraca | Scalpellomorpha | Neolepadidae | MN061491.1 | ||
| Austinogebia edulis (Ngoc-Ho & Chan, 1992) | Malacostraca | Decapoda | Upogebiidae | JN897376.1 | ||
| Typhlatya galapagensis (Monod & Cals, 1970) | Atyidae | KX844711.1 | ||||
| Munidopsis ennell (Benedict, 1902) | Munidopsidae | MH717896.1 | ||||
| Longpotamon parvum (Dai & Song in Dai, Song, Li, Chen, Wang & Hu, 1985) | Potamidae | MN737134.1 | ||||
| Metaplax longipes (Stimpson, 1858) | Varunidae | NC_040976.1 | ||||
| Scyllarides squammosus (H. Milne Edwards, 1837) | Scyllaridae | NC_044425.1 | ||||
| Artemia tibetiana (Abatzopoulos, Zhang & Sorgeloos, 1998) | Branchiopoda | Anostraca | Artemiidae | JQ975177.1 | ||
| Daphnia pulex (Leydig, 1860) | Anomopoda | Daphniidae | NC_000844.1 | |||
| Compete | ||||||
| Lepeophtheirus salmonis (Krøyer, 1837) | Copepoda | Siphonostomatoida | Caligidae | NC_056769.1 | ||
| Pennell sp. (Oken, 1815) | Pennellidae | ON161759.1 | ||||
| Tigriopus japonicus (Mori, 1938) | Harpacticoida | Harpacticidae | AB060648.1 | |||
| Tigriopus kingsejongensis (Park, S. Lee, Cho, Yoon, Y. Lee & W. Lee, 2014) | MK598762.1 | |||||
| Tigriopus californicus (Baker, 1912) | NC_008831.2 | |||||
| Amphiascoides atopus (Lotufo & Fleeger, 1995) | Miraciidae | NC_023783.1 | ||||
| Paracyclopina nana (Smirnov, 1935) | Cyclopoida | Cyclopettidae | EU877959.1 | |||
| Lamproglena chinensis (Yü, 1937) | Lernaeidae | OQ411234 | ||||
| Lamproglena orientalis (Markevich, 1936) | OQ411235 | |||||
| Lernaea cyprinacea (Linnaeus, 1758) | NC_025239.1 | |||||
| Sinergasilus polycolpus (Markevich, 1940) | Ergasilidae | NC_028085.1 | ||||
| Sinergasilus undulatus (Markevich, 1940) | NC_054173.1 | |||||
| Sinergasilus major (Markevich, 1940) | OQ160840 | |||||
| Calanus hyperboreus (Krøyer, 1838) | Calanoida | Calanidae | NC_019627.1 | |||
| Eurytemora affinis (Poppe, 1880) | Temoridae | NC_046694.1 | ||||
| Phyllodiaptomus tunguidus (Shen & Tai, 1964) | Diaptomidae | NC_046743.1 | ||||
| Calanus simillimus (Giesbrecht, 1902) | Calanidae | NC_063666.1 | ||||
| Labidocera rotunda (Mori, 1929) | Pontellidae | NC_064109.1 | ||||
| partial | ||||||
| Caligus rogercresseyi (Boxshall & Bravo, 2000) | Copepoda | Siphonostomatoida | Caligidae | HQ157565.1 | ||
| Caligus clemensi (Parker & Margolis, 1964) | HQ157566.1 | |||||
| Schizopera knabeni (Lang, 1965) | Harpacticoida | Miraciidae | KF667527.1 | |||
| Calanus sinicus (Brodsky, 1962) | Calanoida | Calanidae | GU355641.1 | |||
| Calanus glacialis (Jaschnov, 1955) | MF422146.1 | |||||
| Calanus finmarchicus (Gunnerus, 1770) | MG001887.1 | |||||
| Lovenula raynerae (Suárez-Morales, Wasserman & Dalu, 2015) | Diaptomidae | MH710604.1 |
| Comparison_Tree | K-Score | Concatenate Length |
|---|---|---|
| rrnS | 2.717 | 1096 |
| rrnL | 2.686 | 1704 |
| tRNAs | 1.441 | 1092 |
| nad4L | 1.231 | 406 |
| nad3 | 1.115 | 391 |
| nad6 | 0.951 | 635 |
| cox3 | 0.913 | 825 |
| nad4 | 0.893 | 1438 |
| nad5 | 0.799 | 1890 |
| cox2 | 0.775 | 726 |
| cytb | 0.768 | 1191 |
| atp6 | 0.738 | 720 |
| cox1 | 0.712 | 1589 |
| nad1 | 0.692 | 993 |
| nad2 | 0.624 | 1123 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, J.; Zhou, Z.; Huang, Y.; Feng, J.; Li, W.; Wang, G.; Hua, C. Evolutionary Rates, Divergence Rates, and Performance of Individual Mitochondrial Genes Based on Phylogenetic Analysis of Copepoda. Genes 2023, 14, 1496. https://doi.org/10.3390/genes14071496
He J, Zhou Z, Huang Y, Feng J, Li W, Wang G, Hua C. Evolutionary Rates, Divergence Rates, and Performance of Individual Mitochondrial Genes Based on Phylogenetic Analysis of Copepoda. Genes. 2023; 14(7):1496. https://doi.org/10.3390/genes14071496
Chicago/Turabian StyleHe, Junzong, Zhihao Zhou, Yan Huang, Jinmei Feng, Wenxiang Li, Guitang Wang, and Congjie Hua. 2023. "Evolutionary Rates, Divergence Rates, and Performance of Individual Mitochondrial Genes Based on Phylogenetic Analysis of Copepoda" Genes 14, no. 7: 1496. https://doi.org/10.3390/genes14071496
APA StyleHe, J., Zhou, Z., Huang, Y., Feng, J., Li, W., Wang, G., & Hua, C. (2023). Evolutionary Rates, Divergence Rates, and Performance of Individual Mitochondrial Genes Based on Phylogenetic Analysis of Copepoda. Genes, 14(7), 1496. https://doi.org/10.3390/genes14071496