
Exploring the Micro-Mosaic Landscape of FGFR3 Mutations in the Ageing Male Germline and Their Potential Implications in Meiotic Differentiation

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Testis, Sperm, and Control DNA
2.2. Testis and Sperm DNA Extraction
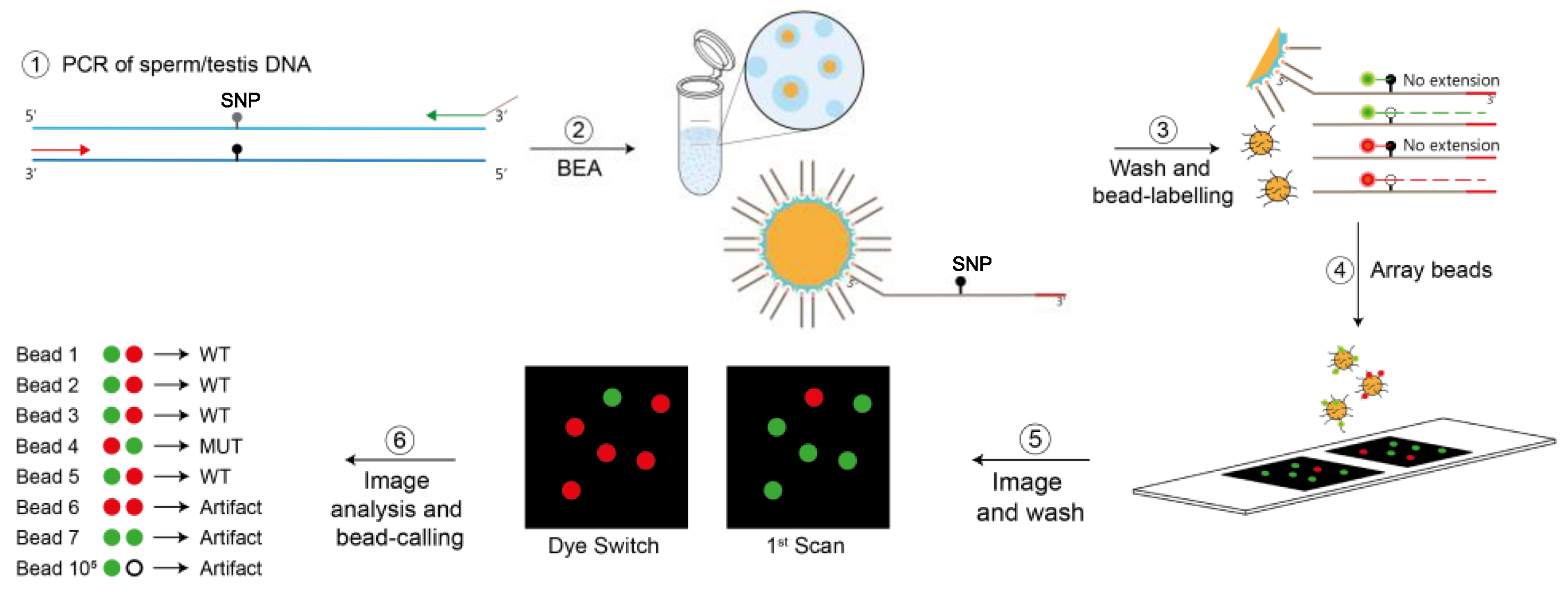
2.3. Bead-Emulsion Amplification (BEA)
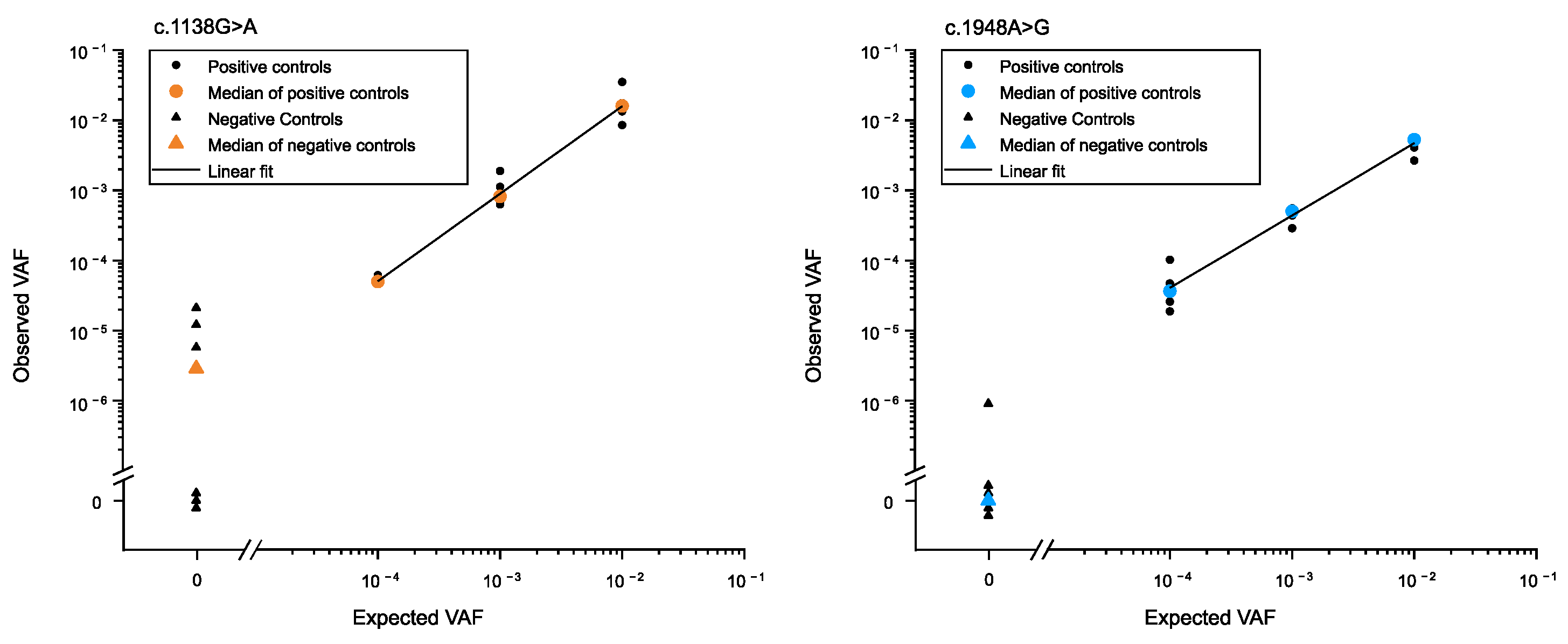
2.4. Statistical Analysis
3. Results

3.1. Determination of Low-Frequency Mutations in the Male Germline
3.2. Transmission of Mutations in Testis
3.3. Transmission of Mutations in Sperm
4. Discussion
4.1. Pathogenic FGFR3 Mutations Accumulate Differently in the Ageing Male Germline
4.2. Activation of Tyrosine Kinase Signalling via the Mutations Associated with ACH and TDII
4.3. Role of RTK-MAPK in Meiotic Differentiation from Sperm into Spermatids
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Horswell, S.; Chambers, T.; O’Brien, T.; Lopez, J.I.; Watkins, T.B.K.; Nicol, D.; et al. Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell 2018, 173, 595–610.e511. [Google Scholar] [CrossRef]
- Yates, L.R.; Gerstung, M.; Knappskog, S.; Desmedt, C.; Gundem, G.; Van Loo, P.; Aas, T.; Alexandrov, L.B.; Larsimont, D.; Davies, H.; et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med. 2015, 21, 751–759. [Google Scholar] [CrossRef]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef]
- Wood, K.A.; Goriely, A. The impact of paternal age on new mutations and disease in the next generation. Fertil. Steril. 2022, 118, 1001–1012. [Google Scholar] [CrossRef]
- Orioli, I.M.; Castilla, E.E.; Barbosa-Neto, J.G. The birth prevalence rates for the skeletal dysplasias. J. Med. Genet. 1986, 23, 328–332. [Google Scholar] [CrossRef]
- Risch, N.; Reich, E.W.; Wishnick, M.M.; McCarthy, J.G. Spontaneous mutation and parental age in humans. Am. J. Hum. Genet. 1987, 41, 218–248. [Google Scholar] [PubMed]
- Crow, J.F. Upsetting the dogma: Germline selection in human males. PLoS Genet. 2012, 8, e1002535. [Google Scholar] [CrossRef] [PubMed]
- Rannan-Eliya, S.V.; Taylor, I.B.; De Heer, I.M.; Van Den Ouweland, A.M.; Wall, S.A.; Wilkie, A.O. Paternal origin of FGFR3 mutations in Muenke-type craniosynostosis. Hum. Genet. 2004, 115, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.; Cordeddu, V.; Chang, H.; Shaw, A.; Kalidas, K.; Crosby, A.; Patton, M.A.; Sorcini, M.; van der Burgt, I.; Jeffery, S.; et al. Paternal germline origin and sex-ratio distortion in transmission of PTPN11 mutations in Noonan syndrome. Am. J. Hum. Genet. 2004, 75, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Zampino, G.; Pantaleoni, F.; Carta, C.; Cobellis, G.; Vasta, I.; Neri, C.; Pogna, E.A.; De Feo, E.; Delogu, A.; Sarkozy, A.; et al. Diversity, parental germline origin, and phenotypic spectrum of de novo HRAS missense changes in Costello syndrome. Hum. Mutat. 2007, 28, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Arnheim, N.; Calabrese, P. Understanding what determines the frequency and pattern of human germline mutations. Nat. Rev. Genet. 2009, 10, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Arnheim, N.; Calabrese, P. Germline Stem Cell Competition, Mutation Hot Spots, Genetic Disorders, and Older Fathers. Annu. Rev. Genom. Hum. Genet. 2016, 17, 219–243. [Google Scholar] [CrossRef]
- Goriely, A.; Wilkie, A.O. Paternal age effect mutations and selfish spermatogonial selection: Causes and consequences for human disease. Am. J. Hum. Genet. 2012, 90, 175–200. [Google Scholar] [CrossRef] [PubMed]
- Goriely, A.; Hansen, R.M.; Taylor, I.B.; Olesen, I.A.; Jacobsen, G.K.; McGowan, S.J.; Pfeifer, S.P.; McVean, G.A.; Rajpert-De Meyts, E.; Wilkie, A.O. Activating mutations in FGFR3 and HRAS reveal a shared genetic origin for congenital disorders and testicular tumors. Nat. Genet. 2009, 41, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Goriely, A.; McVean, G.A.; Rojmyr, M.; Ingemarsson, B.; Wilkie, A.O. Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science 2003, 301, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Tiemann-Boege, I.; Navidi, W.; Grewal, R.; Cohn, D.; Eskenazi, B.; Wyrobek, A.J.; Arnheim, N. The observed human sperm mutation frequency cannot explain the achondroplasia paternal age effect. Proc. Natl. Acad. Sci. USA 2002, 99, 14952–14957. [Google Scholar] [CrossRef]
- Yoon, S.R.; Qin, J.; Glaser, R.L.; Jabs, E.W.; Wexler, N.S.; Sokol, R.; Arnheim, N.; Calabrese, P. The ups and downs of mutation frequencies during aging can account for the Apert syndrome paternal age effect. PLoS Genet. 2009, 5, e1000558. [Google Scholar] [CrossRef]
- Moura, S.; Hartl, I.; Brumovska, V.; Yasari, A.; Striedner, Y.; Bishara, M.; Mair, T.; Ebner, T.; Schütz, G.J.; Calabrese, P.P.; et al. Exploring FGFR3 mutations in the male germline: Implications for clonal germline expansions and paternal age-related dysplasias. Genome Biol. Evol. 2024. accepted. [Google Scholar]
- Choi, S.K.; Yoon, S.R.; Calabrese, P.; Arnheim, N. A germ-line-selective advantage rather than an increased mutation rate can explain some unexpectedly common human disease mutations. Proc. Natl. Acad. Sci. USA 2008, 105, 10143–10148. [Google Scholar] [CrossRef]
- Choi, S.K.; Yoon, S.R.; Calabrese, P.; Arnheim, N. Positive selection for new disease mutations in the human germline: Evidence from the heritable cancer syndrome multiple endocrine neoplasia type 2B. PLoS Genet. 2012, 8, e1002420. [Google Scholar] [CrossRef]
- Eboreime, J.; Choi, S.K.; Yoon, S.R.; Sadybekov, A.; Katritch, V.; Calabrese, P.; Arnheim, N. Germline selection of PTPN11 (HGNC:9644) variants make a major contribution to both Noonan syndrome’s high birth rate and the transmission of sporadic cancer variants resulting in fetal abnormality. Hum. Mutat. 2022, 43, 2205–2221. [Google Scholar] [CrossRef]
- Maher, G.J.; Ralph, H.K.; Ding, Z.; Koelling, N.; Mlcochova, H.; Giannoulatou, E.; Dhami, P.; Paul, D.S.; Stricker, S.H.; Beck, S.; et al. Selfish mutations dysregulating RAS-MAPK signaling are pervasive in aged human testes. Genome Res. 2018, 28, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Calabrese, P.; Tiemann-Boege, I.; Shinde, D.N.; Yoon, S.R.; Gelfand, D.; Bauer, K.; Arnheim, N. The molecular anatomy of spontaneous germline mutations in human testes. PLoS Biol. 2007, 5, e224. [Google Scholar] [CrossRef] [PubMed]
- Shinde, D.N.; Elmer, D.P.; Calabrese, P.; Boulanger, J.; Arnheim, N.; Tiemann-Boege, I. New evidence for positive selection helps explain the paternal age effect observed in achondroplasia. Hum. Mol. Genet. 2013, 22, 4117–4126. [Google Scholar] [CrossRef]
- Yoon, S.R.; Choi, S.K.; Eboreime, J.; Gelb, B.D.; Calabrese, P.; Arnheim, N. Age-dependent germline mosaicism of the most common noonan syndrome mutation shows the signature of germline selection. Am. J. Hum. Genet. 2013, 92, 917–926. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Maher, G.J.; McGowan, S.J.; Giannoulatou, E.; Verrill, C.; Goriely, A.; Wilkie, A.O. Visualizing the origins of selfish de novo mutations in individual seminiferous tubules of human testes. Proc. Natl. Acad. Sci. USA 2016, 113, 2454–2459. [Google Scholar] [CrossRef] [PubMed]
- Hartl, I.; Brumovska, V.; Striedner, Y.; Yasari, A.; Schutz, G.J.; Sevcsik, E.; Tiemann-Boege, I. Measurement of FGFR3 signaling at the cell membrane via total internal reflection fluorescence microscopy to compare the activation of FGFR3 mutants. J. Biol. Chem. 2023, 299, 102832. [Google Scholar] [CrossRef] [PubMed]
- Arbeithuber, B.; Betancourt, A.J.; Ebner, T.; Tiemann-Boege, I. Crossovers are associated with mutation and biased gene conversion at recombination hotspots. Proc. Natl. Acad. Sci. USA 2015, 112, 2109–2114. [Google Scholar] [CrossRef] [PubMed]
- Palzenberger, E.; Reinhardt, R.; Muresan, L.; Palaoro, B.; Tiemann-Boege, I. Discovery of Rare Haplotypes by Typing Millions of Single-Molecules with Bead Emulsion Haplotyping (BEH). Methods Mol. Biol. 2017, 1551, 273–305. [Google Scholar] [CrossRef]
- Boulanger, J.; Muresan, L.; Tiemann-Boege, I. Massively parallel haplotyping on microscopic beads for the high-throughput phase analysis of single molecules. PLoS ONE 2012, 7, e36064. [Google Scholar] [CrossRef]
- Tiemann-Boege, I.; Curtis, C.; Shinde, D.N.; Goodman, D.B.; Tavare, S.; Arnheim, N. Product length, dye choice, and detection chemistry in the bead-emulsion amplification of millions of single DNA molecules in parallel. Anal. Chem. 2009, 81, 5770–5776. [Google Scholar] [CrossRef]
- Accogli, A.; Pacetti, M.; Fiaschi, P.; Pavanello, M.; Piatelli, G.; Nuzzi, D.; Baldi, M.; Tassano, E.; Severino, M.S.; Allegri, A.; et al. Association of achondroplasia with sagittal synostosis and scaphocephaly in two patients, an underestimated condition? Am. J. Med. Genet. A 2015, 167A, 646–652. [Google Scholar] [CrossRef]
- Bellus, G.A.; Hefferon, T.W.; Ortiz de Luna, R.I.; Hecht, J.T.; Horton, W.A.; Machado, M.; Kaitila, I.; McIntosh, I.; Francomano, C.A. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am. J. Hum. Genet. 1995, 56, 368–373. [Google Scholar] [PubMed]
- Georgoulis, G.; Alexiou, G.; Prodromou, N. Achondroplasia with synostosis of multiple sutures. Am. J. Med. Genet. A 2011, 155A, 1969–1971. [Google Scholar] [CrossRef] [PubMed]
- Hafner, C.; van Oers, J.M.; Vogt, T.; Landthaler, M.; Stoehr, R.; Blaszyk, H.; Hofstaedter, F.; Zwarthoff, E.C.; Hartmann, A. Mosaicism of activating FGFR3 mutations in human skin causes epidermal nevi. J. Clin. Investig. 2006, 116, 2201–2207. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, F.; Bonaventure, J.; Legeai-Mallet, L.; Pelet, A.; Rozet, J.M.; Maroteaux, P.; Le Merrer, M.; Munnich, A. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature 1994, 371, 252–254. [Google Scholar] [CrossRef]
- Shiang, R.; Thompson, L.M.; Zhu, Y.Z.; Church, D.M.; Fielder, T.J.; Bocian, M.; Winokur, S.T.; Wasmuth, J.J. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell 1994, 78, 335–342. [Google Scholar] [CrossRef]
- Salazar, R.; Arbeithuber, B.; Ivankovic, M.; Heinzl, M.; Moura, S.; Hartl, I.; Mair, T.; Lahnsteiner, A.; Ebner, T.; Shebl, O.; et al. Discovery of an unusually high number of de novo mutations in sperm of older men using duplex sequencing. Genome Res. 2022, 32, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, E.V.; Lesovoy, D.M.; Goncharuk, S.A.; Goncharuk, M.V.; Hristova, K.; Arseniev, A.S. Structure of FGFR3 transmembrane domain dimer: Implications for signaling and human pathologies. Structure 2013, 21, 2087–2093. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Hristova, K. The physical basis of FGFR3 response to fgf1 and fgf2. Biochemistry 2011, 50, 8576–8582. [Google Scholar] [CrossRef] [PubMed]
- Naski, M.C.; Wang, Q.; Xu, J.; Ornitz, D.M. Graded activation of fibroblast growth factor receptor 3 by mutations causing achondroplasia and thanatophoric dysplasia. Nat. Genet. 1996, 13, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Nardini, E.; Brents, L.A.; Schrock, E.; Ried, T.; Kuehl, W.M.; Bergsagel, P.L. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat. Genet. 1997, 16, 260–264. [Google Scholar] [CrossRef]
- Tavormina, P.L.; Shiang, R.; Thompson, L.M.; Zhu, Y.Z.; Wilkin, D.J.; Lachman, R.S.; Wilcox, W.R.; Rimoin, D.L.; Cohn, D.H.; Wasmuth, J.J. Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat. Genet. 1995, 9, 321–328. [Google Scholar] [CrossRef]
- Wilcox, W.R.; Tavormina, P.L.; Krakow, D.; Kitoh, H.; Lachman, R.S.; Wasmuth, J.J.; Thompson, L.M.; Rimoin, D.L. Molecular, radiologic, and histopathologic correlations in thanatophoric dysplasia. Am. J. Med. Genet. 1998, 78, 274–281. [Google Scholar] [CrossRef]
- Huang, Z.; Chen, H.; Blais, S.; Neubert, T.A.; Li, X.; Mohammadi, M. Structural mimicry of a-loop tyrosine phosphorylation by a pathogenic FGF receptor 3 mutation. Structure 2013, 21, 1889–1896. [Google Scholar] [CrossRef]
- Webster, M.K.; D’Avis, P.Y.; Robertson, S.C.; Donoghue, D.J. Profound ligand-independent kinase activation of fibroblast growth factor receptor 3 by the activation loop mutation responsible for a lethal skeletal dysplasia, thanatophoric dysplasia type II. Mol. Cell Biol. 1996, 16, 4081–4087. [Google Scholar] [CrossRef]
- Bellus, G.A.; Spector, E.B.; Speiser, P.W.; Weaver, C.A.; Garber, A.T.; Bryke, C.R.; Israel, J.; Rosengren, S.S.; Webster, M.K.; Donoghue, D.J.; et al. Distinct missense mutations of the FGFR3 lys650 codon modulate receptor kinase activation and the severity of the skeletal dysplasia phenotype. Am. J. Hum. Genet. 2000, 67, 1411–1421. [Google Scholar] [CrossRef]
- Stephenson, J.D.; Laskowski, R.A.; Nightingale, A.; Hurles, M.E.; Thornton, J.M. VarMap: A web tool for mapping genomic coordinates to protein sequence and structure and retrieving protein structural annotations. Bioinformatics 2019, 35, 4854–4856. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Andersen, P.E., Jr. Prevalence of lethal osteochondrodysplasias in Denmark. Am. J. Med. Genet. 1989, 32, 484–489. [Google Scholar] [CrossRef]
- Bonaventure, J.; Horne, W.C.; Baron, R. The localization of FGFR3 mutations causing thanatophoric dysplasia type I differentially affects phosphorylation, processing and ubiquitylation of the receptor. FEBS J. 2007, 274, 3078–3093. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, L.; Legeai-Mallet, L. FGFR3 intracellular mutations induce tyrosine phosphorylation in the Golgi and defective glycosylation. Biochim. Biophys. Acta 2007, 1773, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Lievens, P.M.; Mutinelli, C.; Baynes, D.; Liboi, E. The kinase activity of fibroblast growth factor receptor 3 with activation loop mutations affects receptor trafficking and signaling. J. Biol. Chem. 2004, 279, 43254–43260. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Grow, E.J.; Mlcochova, H.; Maher, G.J.; Lindskog, C.; Nie, X.; Guo, Y.; Takei, Y.; Yun, J.; Cai, L.; et al. The adult human testis transcriptional cell atlas. Cell Res. 2018, 28, 1141–1157. [Google Scholar] [CrossRef]
- Guo, J.; Nie, X.; Giebler, M.; Mlcochova, H.; Wang, Y.; Grow, E.J.; Kim, R.; Tharmalingam, M.; Matilionyte, G.; Lindskog, C. The Dynamic Transcriptional Cell Atlas of Testis Development during Human Puberty. Cell Stem Cell 2020, 26, 262–276.e264. [Google Scholar] [CrossRef] [PubMed]
- Hermann, B.P.; Cheng, K.; Singh, A.; Roa-De La Cruz, L.; Mutoji, K.N.; Chen, I.C.; Gildersleeve, H.; Lehle, J.D.; Mayo, M.; Westernstroer, B.; et al. The Mammalian Spermatogenesis Single-Cell Transcriptome, from Spermatogonial Stem Cells to Spermatids. Cell Rep. 2018, 25, 1650–1667.e8. [Google Scholar] [CrossRef]
- Puri, P.; Phillips, B.T.; Suzuki, H.; Orwig, K.E.; Rajkovic, A.; Lapinski, P.E.; King, P.D.; Feng, G.S.; Walker, W.H. The transition from stem cell to progenitor spermatogonia and male fertility requires the SHP2 protein tyrosine phosphatase. Stem Cells 2014, 32, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Walker, W.H. The tyrosine phosphatase SHP2 regulates Sertoli cell junction complexes. Biol. Reprod. 2013, 88, 59. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, W.S.; Yi, J.; Kong, S.B.; Ding, J.C.; Zhao, Y.N.; Tian, Y.P.; Feng, G.S.; Li, C.J.; Liu, W.; et al. The role of tyrosine phosphatase Shp2 in spermatogonial differentiation and spermatocyte meiosis. Asian J. Androl. 2020, 22, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Nadarajan, S.; Mohideen, F.; Tzur, Y.B.; Ferrandiz, N.; Crawley, O.; Montoya, A.; Faull, P.; Snijders, A.P.; Cutillas, P.R.; Jambhekar, A.; et al. The MAP kinase pathway coordinates crossover designation with disassembly of synaptonemal complex proteins during meiosis. eLife 2016, 5, e12039. [Google Scholar] [CrossRef]
- Yu, K.; Herr, A.B.; Waksman, G.; Ornitz, D.M. Loss of fibroblast growth factor receptor 2 ligand-binding specificity in Apert syndrome. Proc. Natl. Acad. Sci. USA 2000, 97, 14536–14541. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Disorder | Age of Donor | Mean VAF | Median VAF | IQR | Max VAF | MaxVAF/IQR | n |
|---|---|---|---|---|---|---|---|---|
| c.1138G>A p.G380R | ACH | 68 | 3.5−5 | 2.3−5 | 3.4−5 | 4.1−4 | 12 | 190 |
| %80 | %1.3−4 | 4.0−6 | 4.7−5 | 3.1−3 | 65 | %192 | ||
| c.1948A>G p.K650E | TDII | 68 | 3.4−5 | 0.0 | 6.1−6 | 3.0−3 | 485 | 190 |
| Disorder | Variant | Mean VAF | Median VAF | IQR | Max VAF | n |
|---|---|---|---|---|---|---|
| ACH | c.1138G>A p.G380R | 4.1−5 | 2.9−5 | 4.1−5 | 2.6−4 | 56 |
| TDII | c.1948A>G p.K650E | 4.5−6 | 0 | 6.0−6 | 4.7−5 | 55 |
| Sperm Data [18] | ||||||
| Age group | Average (pmg) | Median | Max | Min | Sample size | % a |
| 19–23 | 4.5 | 2.1 | 34.2 | 0 | 23 | 0% |
| 36–68 | 34.4 | 17.2 | 724.2 | 0 | 168 | 18% |
| 75–80 | 16 | 16 | 31.9 | 0 | 3 | 33% |
| Testis data [21] | ||||||
| Age group | Average (pmg) | Median | Max | Min | Sample size | % a |
| 19–23 | 5 | 1.5 | 16 | 1 | 4 | 0% |
| 36–68 | 358.5 | 270 | 861 | 3 | 6 | 83% |
| 75–80 | 278 | 166 | 621 | 25 | 5 | 80% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Striedner, Y.; Arbeithuber, B.; Moura, S.; Nowak, E.; Reinhardt, R.; Muresan, L.; Salazar, R.; Ebner, T.; Tiemann-Boege, I. Exploring the Micro-Mosaic Landscape of FGFR3 Mutations in the Ageing Male Germline and Their Potential Implications in Meiotic Differentiation. Genes 2024, 15, 191. https://doi.org/10.3390/genes15020191
Striedner Y, Arbeithuber B, Moura S, Nowak E, Reinhardt R, Muresan L, Salazar R, Ebner T, Tiemann-Boege I. Exploring the Micro-Mosaic Landscape of FGFR3 Mutations in the Ageing Male Germline and Their Potential Implications in Meiotic Differentiation. Genes. 2024; 15(2):191. https://doi.org/10.3390/genes15020191
Chicago/Turabian StyleStriedner, Yasmin, Barbara Arbeithuber, Sofia Moura, Elisabeth Nowak, Ronja Reinhardt, Leila Muresan, Renato Salazar, Thomas Ebner, and Irene Tiemann-Boege. 2024. "Exploring the Micro-Mosaic Landscape of FGFR3 Mutations in the Ageing Male Germline and Their Potential Implications in Meiotic Differentiation" Genes 15, no. 2: 191. https://doi.org/10.3390/genes15020191