
Mining Heat-Resistant Key Genes of Peony Based on Weighted Gene Co-Expression Network Analysis
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. The Construction Process of WGCNA
2.2.2. Specific Module Screening
2.2.3. Interaction Network Prediction of Protein−Transcription Factor and Key Gene Mining
3. Results
3.1. Construction and Module Division of Gene Co−Expression Network
3.2. Specific Module Screening
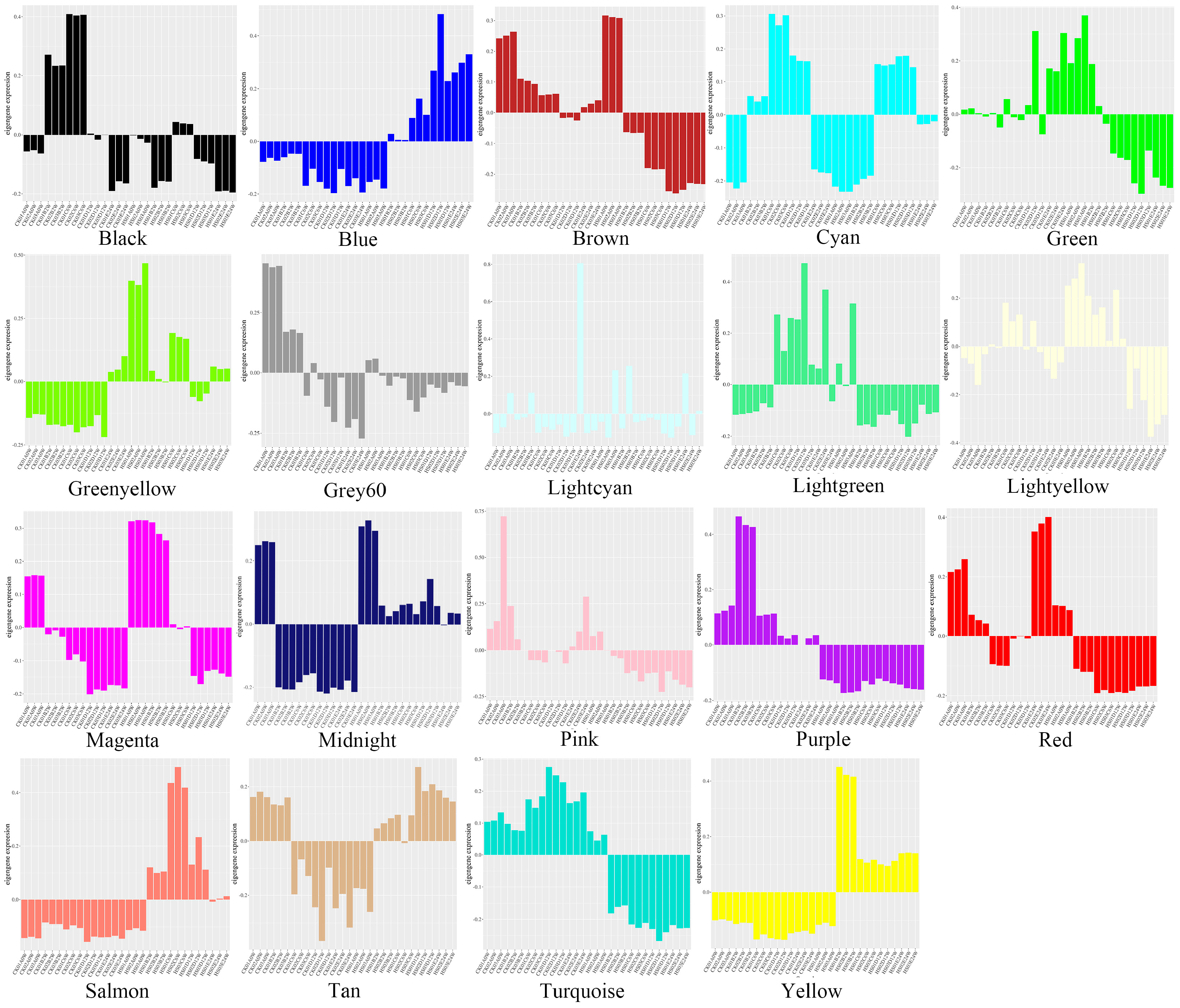
3.2.1. Analysis of Module Characteristic Gene Expression Pattern
3.2.2. GO and KEGG Enrichment Analysis of Module Characteristic Genes
3.2.3. The Gene Expression of Each Module between Different Treatments
3.3. Prediction of Gene−Protein Interaction Network of Main Functional Modules and Mining of Key Genes
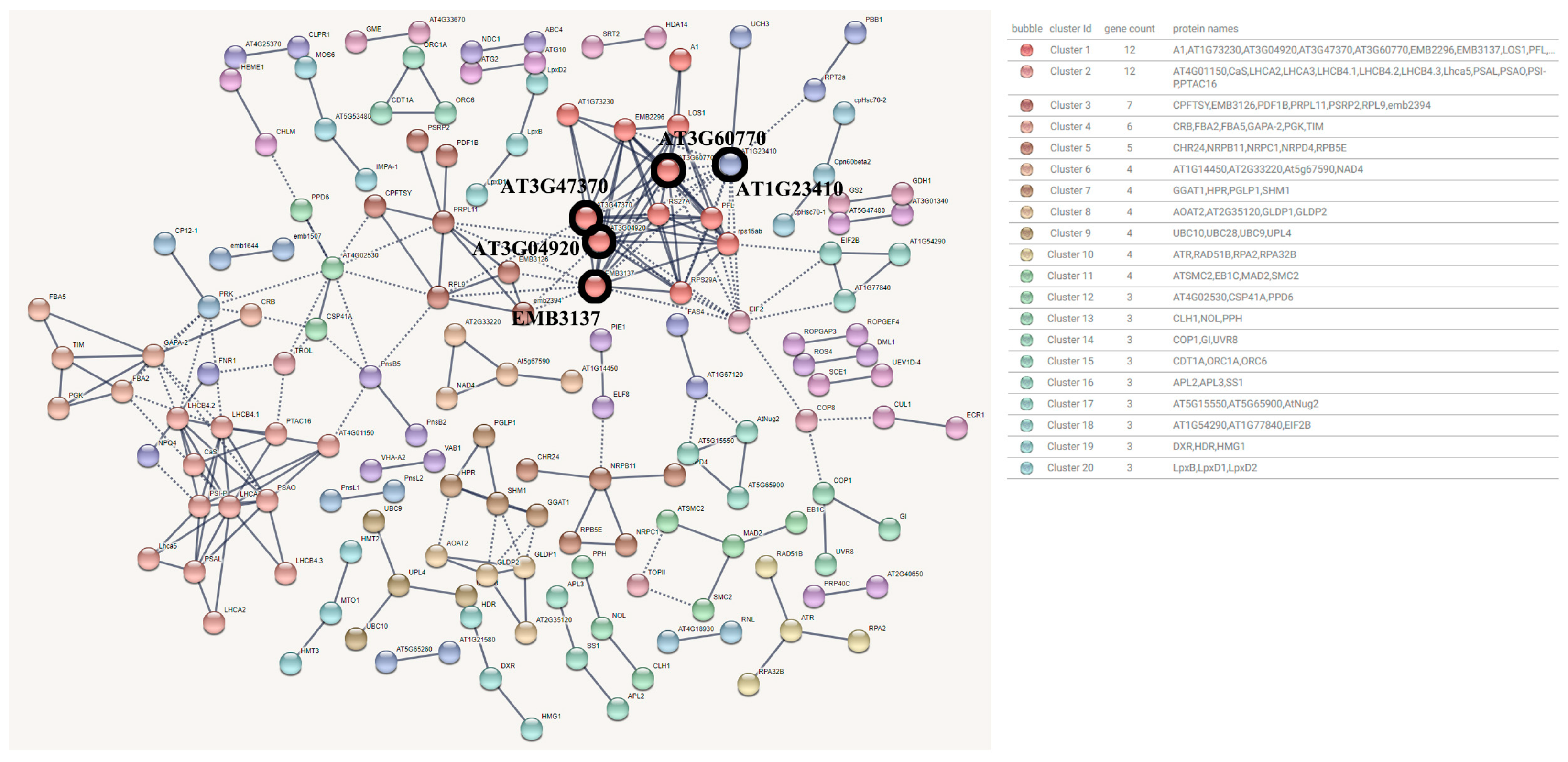
3.3.1. Blue Module Protein Interaction Network
3.3.2. Brown Module Protein Interaction Network
3.3.3. Yellow Module Protein Interaction Network
3.4. Prediction and Analysis of Transcription Factor Interaction Network of Main Modules
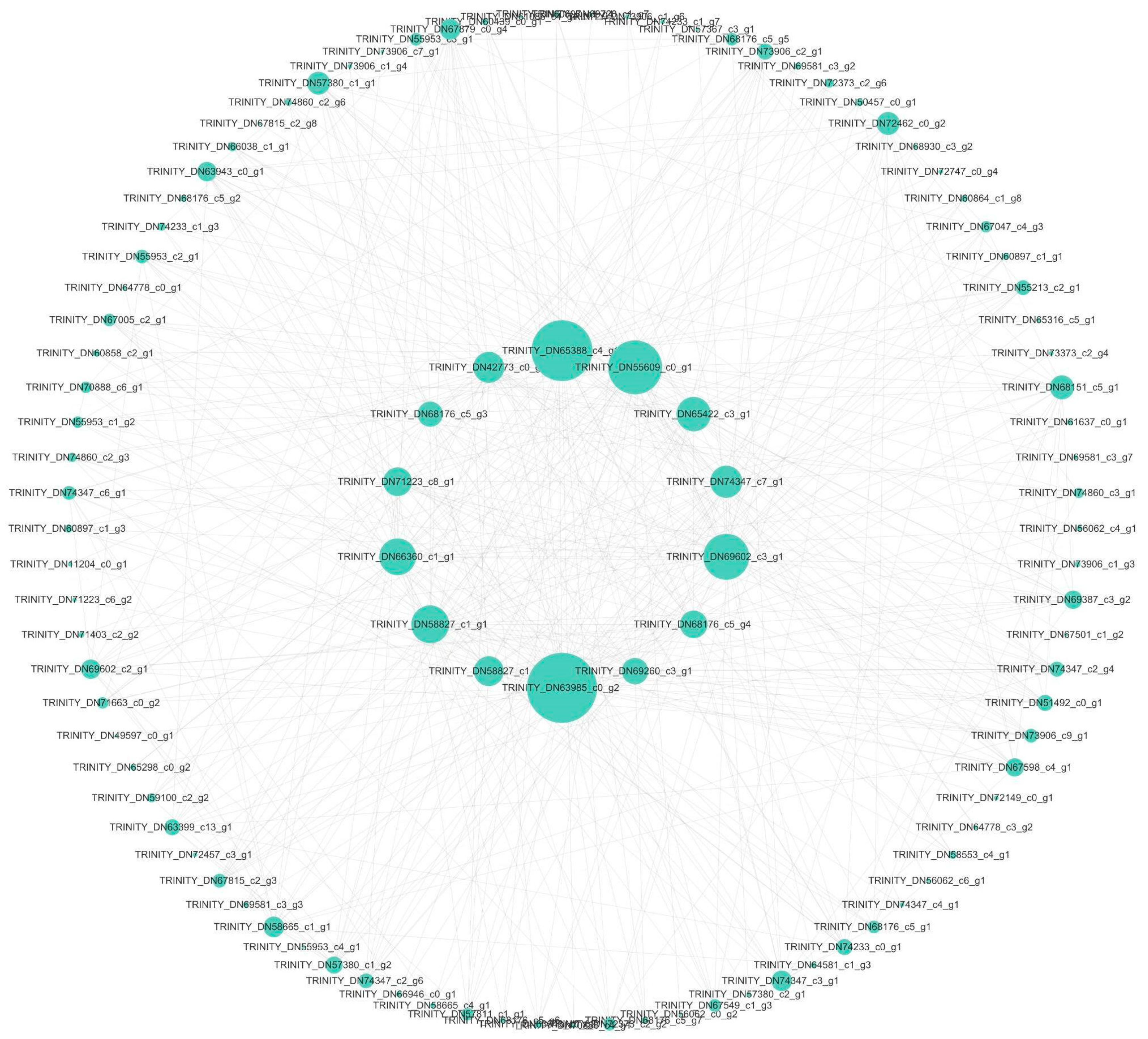
3.4.1. Blue Module Transcription Factor Interaction Network
3.4.2. Brown Module Transcription Factor Interaction Network
3.4.3. Yellow Module Transcription Factor Interaction Network
4. Discussion
- WGCNA is considered to be an efficient gene mining technology. It can specifically screen out the relevant genes that meet the requirements through the modular classification method, thereby obtaining a highly relevant co-expression module [26]. Zhang et al. [27] used the WGCNA method to divide and analyze the specific modules, and successfully excavated six candidate genes related to foxtail millet cold stress. Deng et al. [28] used WGCNA co-expression network analysis to predict 10 abiotic stress core genes in maize, and these genes may be the core genes of the abiotic stress module. Based on WGCNA analysis, Li et al. [29] excavated five key genes related to the anaerobic germination of rice as a second item.
- In the WGCNA network module, those genes that connect a large number of genes and have a high degree of connectivity are called hub genes. These hub genes play a crucial role in the network [30]. Through cluster analysis, it was determined that there was a high co-expression relationship between WRKY33, WRKY40 and WRKY53 transcription factors. Studies have shown that WRKY33 and WRKY40 can negatively regulate salicylic acid and jasmonic acid signaling pathways and biosynthesis [31]. However, the expression of WRKY33 and WRKY40 was not affected by high temperature stress, while the expression of PsWRKY53 was significantly different. It can be seen that PsWRKY53 may activate the key factors of the ethylene signaling pathway by co-expressing with PsERF6 and responding to the ethylene response element binding factor PsERF1A and the ethylene transcription factor PsERF11, thereby regulating the heat resistance defense response of peony. Using high-confidence screening of the Yel-low module, it was found that there was a strong co-expression relationship between AtHsfB2b and MBF1C proteins in cluster3. GO enrichment analysis showed that the genes of the Yellow module were highly enriched in the category of ‘ response to heat ‘. And it has been confirmed that the transcription factor can negatively regulate Arabidopsis thermomorphogenesis [32]. Therefore, it is speculated that PsHSFB2b may be a key factor for peony to resist heat stress.
- The key modules of the peony heat shock response were related to the functions of the Blue, Brown, and Yellow module. We predicted that PsWRKY53 (TRINITY_DN60998_c1_g2, TRINITY_DN71537_c0_g1) and PsHsfB2b (TRINITY_DN56794_c0_g1) may play an important role in tree peony under high temperature stress. The WRKY53 transcription factor is mainly involved in plant endogenous hormone pathways. For example, the transcription factor was found to enhance plant stress resistance by regulating the gibberellin, jasmonic acid, and salicylic acid signaling pathways [33,34,35]. And WRKY53 has been reported to regulate plant growth and development, such as regulating the early and middle development of Arabidopsis leaves [36], leaf senescence [37,38] and so on. It can be seen that the WRKY53 transcription factor is widely studied in Arabidopsis thaliana, and the current research has not yet involved thermal response and response to ethylene signaling pathways. Therefore, whether PsWRKY53 responds to ethylene signaling pathway and improves the heat resistance of peony needs further study. Previous studies have shown that HSFB2b plays an important role in plant response to stress conditions such as high temperature, salt, and drought stress [39]. As a transcriptional repressor, HSFB2b in Arabidopsis can regulate the thermomorphogenesis of Arabidopsis by inhibiting the expression of downstream heat-responsive genes at high temperature [40]. However, HSFB2b plays a positive regulatory role in pansy [41]. In this study, PsHSFB2b is located in the Yellow module, which is an expression pattern that first decreases and then increases. How to regulate the thermal response of red peony and its relationship with MBF1C protein needs further verification.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo, J.; Huang, Y.; Yang, X.; Bu, W.; Tian, J.; Zhang, M.; Huang, K.; Luo, X.; Ye, Y.; Xing, W.; et al. Effects of Three Exogenous Substances on Heat Tolerance of Peony Seedlings. Horticulturae 2023, 9, 765. [Google Scholar] [CrossRef]
- Liu, Y.; Xiao, Y. Study on the Application of Narrative Design of Cut Porcelain Carving in Southern Fujian in Modern Furniture Decoration. Furnit. Inter. Des. 2022, 29, 22–29. [Google Scholar]
- Dou, L.; Gong, Y. Design and Application of Clothing Patterns of Women in Yongle Palace in Cultural and Creative Products. Furnit. Inter. Des. 2022, 29, 54–57. [Google Scholar]
- Jia, W. Construction of the Spatial Space for Dining of Dowager Cixi—A Case Study on the Palace of Harmonious Conduct in Forbidden City. Furnit. Inter. Des. 2023, 30, 18–22. [Google Scholar]
- Cao, Y. Studies on History of Cultivation, Species Resources of Hunan Tree Peony and its Application in Gardens. Master’s Thesis, Central South University of Forestry and Technology, Changsha, China, 2012. [Google Scholar]
- Zhang, M.H. The Research on Genetic Diversity and Heat Tolerance of Peony Resources in Hunan Province. Master’s Thesis, Central South University of Forestry and Technology, Changsha, China, 2019. [Google Scholar]
- Yu, T.; Zhang, J.; Cao, J.; Ma, X.; Li, W.; Yang, G. Hub Gene Mining and Co−expression Network Construction of Low-Temperature Response in Maize of Seedling by WGCNA. Genes 2023, 14, 1598. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinf. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J. Studies on the Origin of Chinese Mudan (Tree Peony). J. Beijing For. Univ. 1998, 20, 26–30. [Google Scholar]
- Chen, F.; Yang, X.; Ma, Y.; Wang, X.; Luo, D.; Yang, L.; Yang, Y. Synthesis and application of novel silver magnetic amino silicone adhesive particles for preparation of high purity α-linolenic acid from tree peony seed oil under applied magnetic field. J. Chromatogr. A 2020, 1610, 460540. [Google Scholar] [CrossRef] [PubMed]
- Teixeira da Silva, J.; Shen, M.; Yu, X. Tissue culture and micropropagation of tree peony (Paeonia suffruticosa Andr.). J. Crop Sci. Biotechnol. 2012, 15, 159–168. [Google Scholar] [CrossRef]
- Liu, C.; Chen, D.; Gai, S.; Zhang, Y.; Zheng, G. Effects of high-and low temperature stress on the leaf PSII functions and physiological characteristics of tree peony (Paeonia suffruticosa cv.‘Roufurong’. Chin. J. Appl. Ecol. 2012, 23, 133–139. [Google Scholar]
- von Mering, C.; Huynen, M.; Jaeggi, D.; Schmidt, S.; Bork, P.; Snel, B. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 2003, 31, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Satuluri, V.; Parthasarathy, S.; Ucar, D. Markov clustering of protein interaction networks with improved balance and scalability. In Proceedings of the First ACM International Conference on Bioinformatics and Computational Biology (BCB ‘10), New York, NY, USA, 2 August 2010. [Google Scholar]
- Flematti, G.R.; Dixon, K.W.; Smith, S.M. What are karrikins and how were they ‘discovered’ by plants? BMC Biol. 2015, 13, 108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.M. Preliminary Study on Auxiliary Molecular Chaperones Bag6 and Hop in Arabidopsis Thaliana. Master’s Thesis, Shandong Normal University, Jinan, China, 2018. [Google Scholar]
- Yabuta, Y.; Nishizawa-Yokoi, A.; Ono, K.; Shigeoka, S. Arabidopsis Sgt1a as an important factor for the acquirement of thermotolerance. Plant Sci. 2009, 177, 676–681. [Google Scholar] [CrossRef]
- Zhu, S.; Ma, J.; Hao, L.; Zhang, L. Identification and Analysis of Heat-resistant Differential Protein in Leaves of Paeonia Suffruticosa. Mol. Breed. 2021, 19, 419–431. [Google Scholar]
- Birkenbihl, R.P.; Kracher, B.; Roccaro, M.; Somssich, I.E. Induced Genome-Wide Binding of Three Arabidopsis WRKY Transcription Factors during Early MAMP-Triggered Immunity. Plant Cell 2017, 29, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Dubois, M.; Van den Broeck, L.; Claeys, H.; Van Vlierberghe, K.; Matsui, M.; Inzé, D. The ETHYLENE RESPONSE FACTORs ERF6 and ERF11 Antagonistically Regulate Mannitol-Induced Growth Inhibition in Arabidopsis. Plant Physiol. 2015, 169, 166–179. [Google Scholar] [CrossRef]
- Wang, W.; Ryu, K.H.; Bruex, A.; Barron, C.; Schiefelbein, J. Molecular basis for a cell fate switch in re-sponse to impaired ribosome biogenesis in the Arabidopsis root epidermis. Plant Cell 2020, 32, 2402–2423. [Google Scholar] [CrossRef]
- Petroni, K.; Kumimoto, R.W.; Gnesutta, N.; Calvenzani, V.; Fornari, M.; Tonelli, C.; Holt, B.F.; Mantovani, R. The promiscuous life of plant NUCLEAR FACTOR Y transcription factors. Plant Cell 2012, 24, 4777–4792. [Google Scholar] [CrossRef]
- Zhiponova, M.K.; Morohashi, K.; Vanhoutte, I.; Machemer-Noonan, K.; Revalska, M.; Van Montagu, M.; Grotewold, E.; Russinova, E. Helix-loop-helix/basic helix-loop-helix transcription factor network represses cell elongation in Arabidopsis through an apparent incoherent feed-forward loop. Proc. Natl. Acad. Sci. USA 2014, 111, 2824–2829. [Google Scholar] [CrossRef]
- Yu, B.; Tian, Y.; Li, H.; Lu, X.; Wang, Y.; Duanmu, H. Research Progress of Plant bHLH Transcription Factor. Chin. Agric. Sci. Bull. 2019, 35, 75–80. [Google Scholar]
- Li, S.; Fu, Q.; Huang, W.; Yu, D. Functional analysis of an Arabidopsis transcription factor WRKY25 in heat stress. Plant Cell Rep. 2009, 28, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Liu, Y.; Qiu, L.; Zhou, M.; Du, X.; Dai, S.; Sun, M. Advance in Molecular Mechanism of MBF1 Regulating Plant Heat Response and Development. Chin. Bull. Bot. 2022, 57, 56–68. [Google Scholar]
- Zhao, W.; Langfelder, P.; Fuller, T.; Dong, J.; Li, A.; Hovarth, S. Weighted gene coexpression network analysis: State of the art. J. Biopharm. Stat. 2010, 20, 281–300. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.; Zhao, B.; Zhang, L.; Qie, Q.; Han, Y.; Li, X. Identification of Co-expression Genes Related to Cold Stress in Foxtail Millet by WGCNA. J. Agric. Sci. Technol. 2023, 25, 22–34. [Google Scholar]
- Deng, Z.; Jiang, H.; Chen, L.; Liu, R.; Huang, M.; Li, M.; Du, H. Identification of abiotic stress-related gene co-expression networks in maize by WGCNA. Acta Agron. Sin. 2023, 49, 672–686. [Google Scholar]
- Li, D.; Liu, K.; Zhao, C.; Liang, S.; Yang, J.; Peng, Z.; Xia, A.; Yang, M.; Luo, L.; Huang, C.; et al. GWAS Combined with WGCNA of Transcriptome and Metabolome to Ex-cavate Key Candidate Genes for Rice Anaerobic Germination. Rice 2023, 16, 49. [Google Scholar] [CrossRef]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef]
- Najafi, J.; Brembu, T.; Vie, A.K.; Viste, R.; Winge, P.; Somssich, I.E.; Bones, A.M. PAMP-INDUCED SECRETED PEPTIDE 3 modulates immunity in Arabidopsis. J. Exp. Bot. 2020, 71, 850–864. [Google Scholar]
- Yao, Z.; Sun, J.; Liu, J.; Lu, H. Heat shock transcription factor HSFB2b negatively regulates plant thermomorphogenesis in Arabidopsis. J. Zhejiang Univ. 2023, 49, 23–30. [Google Scholar]
- Tang, J.; Tian, X.; Mei, E.; He, M.; Gao, J.; Yu, J.; Xu, M.; Liu, J.; Song, L.; Li, X.; et al. WRKY53 negatively regulates rice cold tolerance at the booting stage by fine-tuning anther gibberellin levels. Plant Cell 2022, 34, 4495–4515. [Google Scholar] [CrossRef]
- Jiao, C.; Li, K.; Zuo, Y.; Gong, J.; Guo, Z.; Shen, Y. CALMODULIN1 and WRKY53 Function in Plant Defense by Neg-atively Regulating the Jasmonic Acid Biosynthesis Pathway in Arabidopsis. Int. J. Mol. Sci. 2022, 23, 7718. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Das, A.; Khandagale, P.; Maiti, I.B.; Chattopadhyay, S.; Dey, N. Interaction of Arabidopsis TGA3 and WRKY53 transcription factors on Cestrum yellow leaf curling virus (CmYLCV) promoter mediates salicylic ac-id-dependent gene expression in planta. Planta 2018, 247, 181–199. [Google Scholar] [CrossRef]
- Xie, Y.; Huhn, K.; Brandt, R.; Potschin, M.; Bieker, S.; Straub, D.; Doll, J.; Drechsler, T.; Zentgraf, U.; Wenkel, S. REVOLUTA and WRKY53 connect early and late leaf development in Arabidopsis. Development 2014, 141, 4772–4783. [Google Scholar] [CrossRef]
- Wang, Q.; Li, X.; Guo, C.; Wen, L.; Deng, Z.; Zhang, Z.; Li, W.; Liu, T.; Guo, Y. Senescence-related receptor kinase 1 functions downstream of WRKY53 in regulating leaf senescence in Arabidopsis. J. Exp. Bot. 2023, 74, 5140–5152. [Google Scholar] [CrossRef]
- Li, G.H. Function and molecular mechanism of heat shock transcription factor HsfB1 and HsfB2b in abiotic stress. Master’s Thesis, Henan Agricultural University, Zhengzhou, China, 2022. [Google Scholar]
- Ikeda, M.; Mitsuda, N.; Ohme-Takagi, M. Arabidopsis HsfB1 and HsfB2b act as repressors of the expression of heat-inducible Hsfs but positively regulate the acquired thermotolerance. Plant Physiol. 2011, 157, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Zhu, X.; Du, X.; Liu, H. Cloning of HSFB2b Gene from Pansy and Analysis on Its Expression to Heat Stress. Mol. Plant Breed. 2021, 19, 780–785. [Google Scholar]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Huang, Y.; Yao, Y.; Bu, W.; Zhang, M.; Zheng, T.; Luo, X.; Wang, Z.; Lei, W.; Tian, J.; et al. Mining Heat-Resistant Key Genes of Peony Based on Weighted Gene Co-Expression Network Analysis. Genes 2024, 15, 383. https://doi.org/10.3390/genes15030383
Yang X, Huang Y, Yao Y, Bu W, Zhang M, Zheng T, Luo X, Wang Z, Lei W, Tian J, et al. Mining Heat-Resistant Key Genes of Peony Based on Weighted Gene Co-Expression Network Analysis. Genes. 2024; 15(3):383. https://doi.org/10.3390/genes15030383
Chicago/Turabian StyleYang, Xingyu, Yu Huang, Yiping Yao, Wenxuan Bu, Minhuan Zhang, Tangchun Zheng, Xiaoning Luo, Zheng Wang, Weiqun Lei, Jianing Tian, and et al. 2024. "Mining Heat-Resistant Key Genes of Peony Based on Weighted Gene Co-Expression Network Analysis" Genes 15, no. 3: 383. https://doi.org/10.3390/genes15030383