
Identification of Crucial Modules and Genes Associated with Bt Gene Expression in Cotton
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Bt Protein Content Analysis
2.2. RNA Sequencing Data Generation and Processing
2.3. Weighted Co-Expression Network Construction
2.4. Virus-Induced Gene Silencing of Hub Genes
2.5. Quantitative Reverse-Transcription PCR
2.6. Statistical Analysis
3. Results
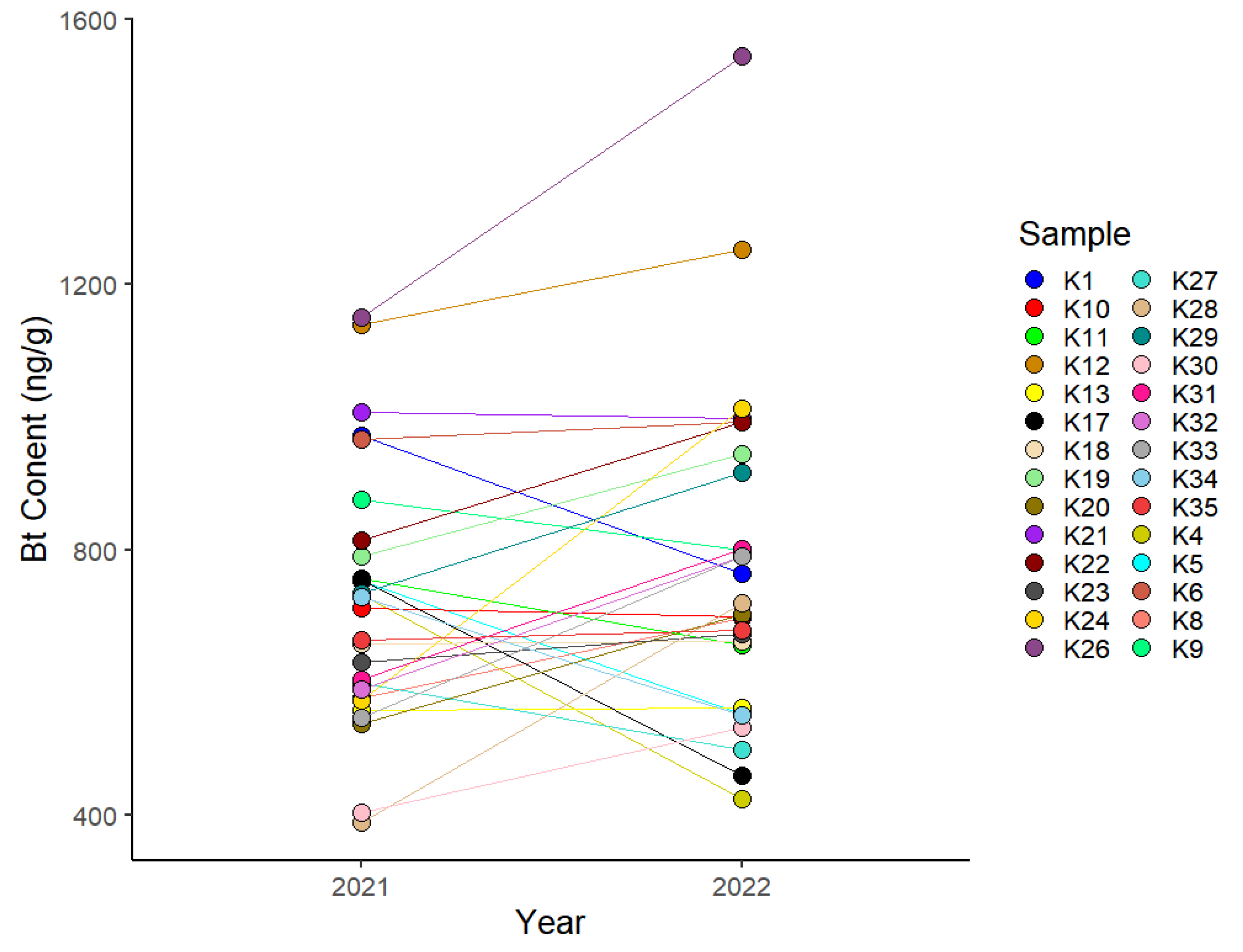
3.1. Quantification of Cry1Ac Bt Toxin
3.2. RNA-Seq Data and Analysis
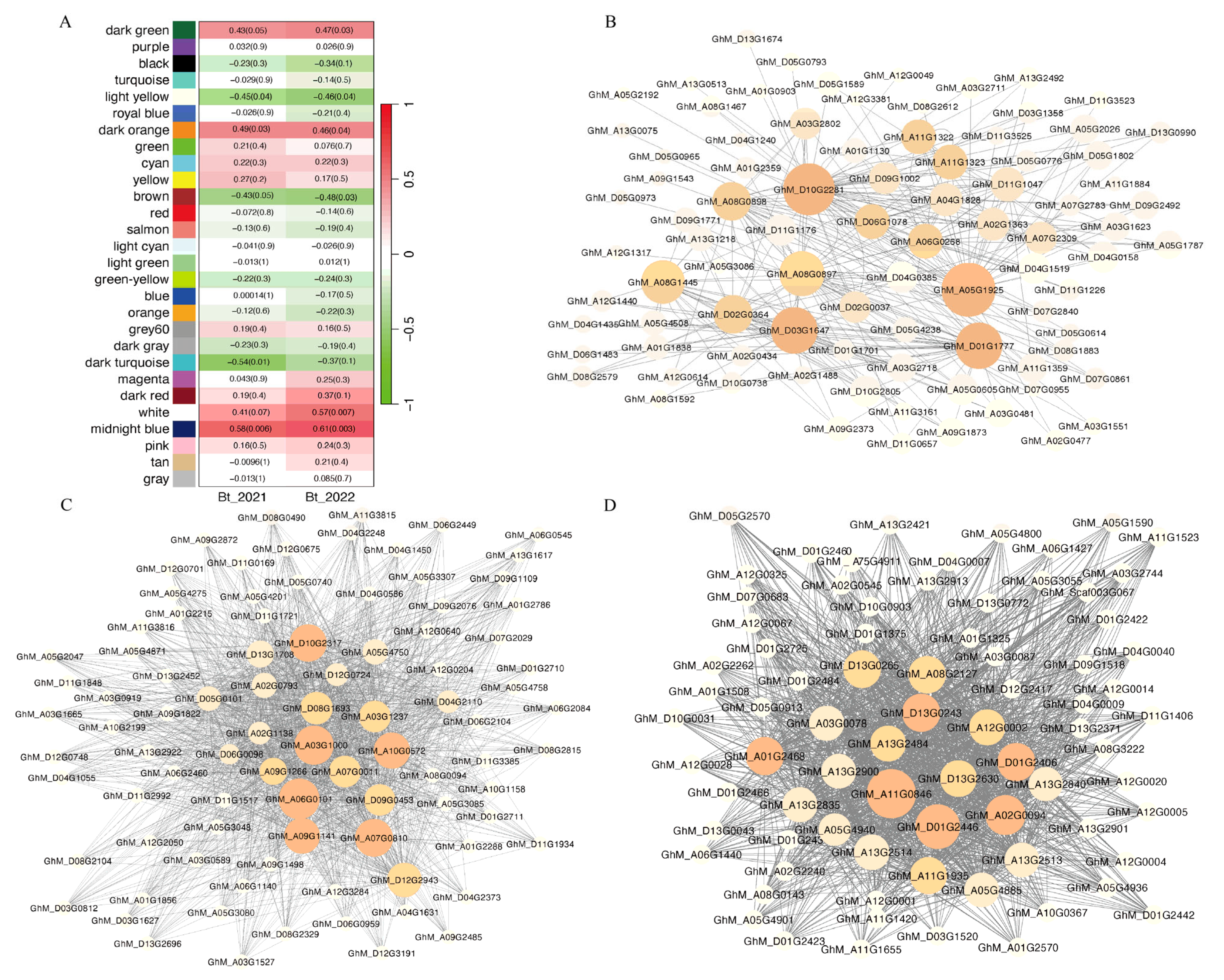
3.3. Identification of Crucial Modules
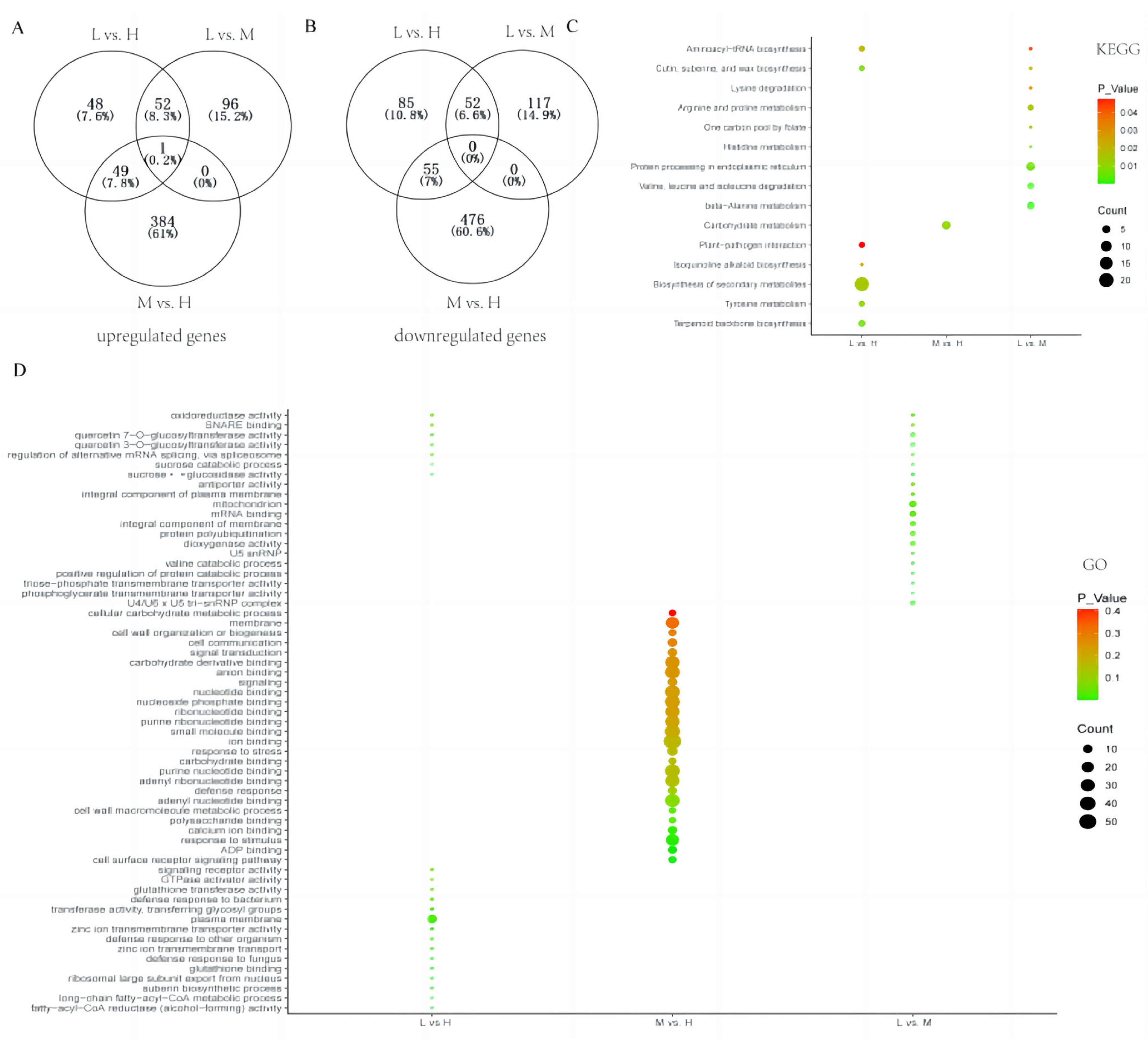
3.4. Functional Enrichment Analysis of the Genes in the Crucial Modules
3.5. Identification of the Hub Gene Associated with Bt Protein Content
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barwale, R.B. Prospects for Bt Cotton Technology in India. Agbioforum 2004, 7, 23–26. [Google Scholar]
- Dong, H.Z.; Li, W.J.; Tang, W.; Li, Z.H.; Zhang, D.M. Increased Yield and Revenue with a Seedling Transplanting System for Hybrid Seed Production in Bt Cotton. J. Agron. Crop Sci. 2005, 191, 116–124. [Google Scholar] [CrossRef]
- Smitha, P.K.; Bathula, C.; Kumar, A.M.; Chandrashekara, K.N.; Dhar, S.K.; Das, M. Correlation of Cry1Ac mRNA and protein abundance in transgenic Gossypium hirsutum plant. 3 Biotech 2021, 11, 289. [Google Scholar] [CrossRef] [PubMed]
- Ping, S.; Kejian, L.; Yongjun, Z.; Kongming, W.; Yuyuan, G. Seasonal Expression of Bacillus thuringiensis Insecticidal Protein and the Control to Cotton Bollworm in Different Varieties of Transgenic Cotton. Cotton Sci. 2010, 22, 393–397. [Google Scholar]
- Mahon, R.J.F.K. Environmental stress and the efficacy of Bt cotton. Aust. Cotton Grower 2002, 22, 18–21. [Google Scholar]
- Guo, W.Z.; Sun, J.; Guo, Y.F.; Zhang, T.Z. Investigation of different dosages of inserted Bt genes and their insect-resistance in transgenic Bt cotton. Yi Chuan Xue Bao 2001, 28, 668–676. [Google Scholar] [PubMed]
- Lan-Qin, X.; Qiong-Fang, X.U.; San-Dui, G. Bt Insecticidal Gene and Its Temporal Expression in Transgenic Cotton Plants. Acta Agron. Sin. 2005, 31, 197–202. [Google Scholar]
- Bruns, H.A.; Abel, C.A. Nitrogen Fertility Effects on Bt δ-Endotoxin and Nitrogen Concentrations of Maize during Early Growth. Agron. J. 2003, 95, 207–211. [Google Scholar]
- Coviella, C.E. Plant allocation to defensive compounds: Interactions between elevated CO2 and nitrogen in transgenic cotton plants. J. Exp. Bot. 2002, 53, 323–331. [Google Scholar] [CrossRef]
- Adamczyk, J.J.; Meredith, W.R. Genetic Basis for Variability of Cry1Ac Expression Among Commercial Transgenic B. thuringiensis (Bt) Cotton Cultivars in the United States. J. Cotton Sci. 2004, 8, 17–23. [Google Scholar]
- Cheema, H.M.N.; Khan, A.A.; Khan, M.I.; Aslam, U.; Rana, I.A.; Khan, I.A. Assessment of Bt cotton genotypes for the Cry1Ac transgene and its expression. J. Agric. Sci. 2016, 154, 109–117. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Zheng, Y.; Teixeira Da Silva, J.A.; Yu, X. Comparative transcriptomics and WGCNA reveal candidate genes involved in petaloid stamens in Paeonia lactiflora. J. Hortic. Sci. Biotechnol. 2021, 96, 588–603. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, Y.; Shi, H.; Hu, H.; Yi, L.; Hou, J.; Singh, A.K. Time-course transcriptome and WGCNA analysis revealed the drought response mechanism of two sunflower inbred lines. PLoS ONE 2022, 17, e265447. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Zhao, G.; Geng, Z.; Qi, H.; Dou, H.; Zhang, H. Dynamic transcriptome and co-expression network analysis of the cotton (Gossypium hirsutum) root response to salinity stress at the seedling stage. Acta Physiol. Plant 2020, 42, 143. [Google Scholar] [CrossRef]
- Guo, J.; Gao, J.; Liu, Z. HISAT2 Parallelization Method Based on Spark Cluster. J. Phys. Conf. Ser. 2022, 2179, 12038. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Pettigrew, W.T.; Adamczyk, J.J. Nitrogen Fertility and Planting Date Effects on Lint Yield and Cry1Ac (Bt) Endotoxin Production. Agron. J. 2006, 98, 691–697. [Google Scholar] [CrossRef]
- Jiang, L.; Duan, L.; Tian, X.; Wang, B.; Zhang, H.; Zhang, M.; Li, Z. NaCl salinity stress decreased B. thuringiensis (Bt) protein content of transgenic Bt cotton (Gossypium hirsutum L.) seedlings. Environ. Exp. Bot. 2006, 55, 315–320. [Google Scholar] [CrossRef]
- Chen, D.; Ye, G.; Yang, C.; Chen, Y.; Wu, Y. The effect of high temperature on the insecticidal properties of Bt Cotton. Environ. Exp. Bot. 2005, 53, 333–342. [Google Scholar] [CrossRef]
- Benedict, J.H.; Sachs, E.S.; Altman, D.W.; Deaton, W.R.; Kohel, R.J.; Ring, D.R.; Berberich, S.A. Field performance of cottons expressing transgenic CryIA insecticidal proteins for resistance to Heliothis virescens and Helicoverpa zea (Lepidoptera: Noctuidae). J. Econ. Entomol. 1996, 89, 230–238. [Google Scholar] [CrossRef]
- Du, M.; Zhao, J.; Tzeng, D.T.W.; Liu, Y.; Li, C. MYC2 Orchestrates a Hierarchical Transcriptional Cascade That Regulates Jasmonate-Mediated Plant Immunity in Tomato. Plant Cell 2017, 29, 1883–1906. [Google Scholar] [CrossRef] [PubMed]
- Uji, Y.; Taniguchi, S.; Tamaoki, D.; Shishido, H.; Akimitsu, K.; Gomi, K. Overexpression of OsMYC2 Results in the Up-Regulation of Early JA-Rresponsive Genes and Bacterial Blight Resistance in Rice. Plant Cell Physiol. 2016, 57, 1814–1827. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.S.; Joo, J.; Kim, M.J.; Kim, Y.K.; Nahm, B.H.; Song, S.I.; Cheong, J.J.; Lee, J.S.; Kim, J.K.; Choi, Y.D. OsbHLH148, a basic helix-loop-helix protein, interacts with OsJAZ proteins in a jasmonate signaling pathway leading to drought tolerance in rice. Plant J. 2011, 65, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Min, D.; Li, F.; Zhang, X.; Cui, X.; Shu, P.; Dong, L.; Ren, C. SlMYC2 Involved in Methyl Jasmonate-Induced Tomato Fruit Chilling Tolerance. J. Agric. Food Chem. 2018, 66, 3110–3117. [Google Scholar] [CrossRef]
- Dombrecht, B.; Xue, G.P.; Sprague, S.J.; Kirkegaard, J.A.; Ross, J.J.; Reid, J.B.; Fitt, G.P.; Sewelam, N.; Schenk, P.M.; Manners, J.M.; et al. MYC2 Differentially Modulates Diverse Jasmonate-Dependent Functions in Arabidopsis. Plant Cell 2007, 19, 2225–2245. [Google Scholar] [CrossRef]
- Wang, H.; Li, Y.; Pan, J.; Lou, D.; Hu, Y.; Yu, D. The bHLH transcription factors MYC2, MYC3, and MYC4 are required for jasmonate-mediated inhibition of flowering in Arabidopsis. Mol. Plant 2017, 10, 1461–1464. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, J.; Xie, Z.; Gao, J.; Ren, G.; Gao, S.; Zhou, X.; Kuai, B. Jasmonic acid promotes degreening via MYC2/3/4-and ANAC 019/055/072-mediated regulation of major chlorophyll catabolic genes. Plant J. 2015, 84, 597–610. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, G.; Geng, Z.; Liu, J.; Tian, H.; Liu, X.; An, Z.; Zhao, N.; Zhang, H.; Wu, L.; Wang, X.; et al. Identification of Crucial Modules and Genes Associated with Bt Gene Expression in Cotton. Genes 2024, 15, 515. https://doi.org/10.3390/genes15040515
Zhao G, Geng Z, Liu J, Tian H, Liu X, An Z, Zhao N, Zhang H, Wu L, Wang X, et al. Identification of Crucial Modules and Genes Associated with Bt Gene Expression in Cotton. Genes. 2024; 15(4):515. https://doi.org/10.3390/genes15040515
Chicago/Turabian StyleZhao, Guiyuan, Zhao Geng, Jianguang Liu, Haiyan Tian, Xu Liu, Zetong An, Ning Zhao, Hanshuang Zhang, Liqiang Wu, Xingfen Wang, and et al. 2024. "Identification of Crucial Modules and Genes Associated with Bt Gene Expression in Cotton" Genes 15, no. 4: 515. https://doi.org/10.3390/genes15040515