
SNP and Structural Study of the Notch Superfamily Provides Insights and Novel Pharmacological Targets against the CADASIL Syndrome and Neurodegenerative Diseases
 , , , , ,
, , , , ,  , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
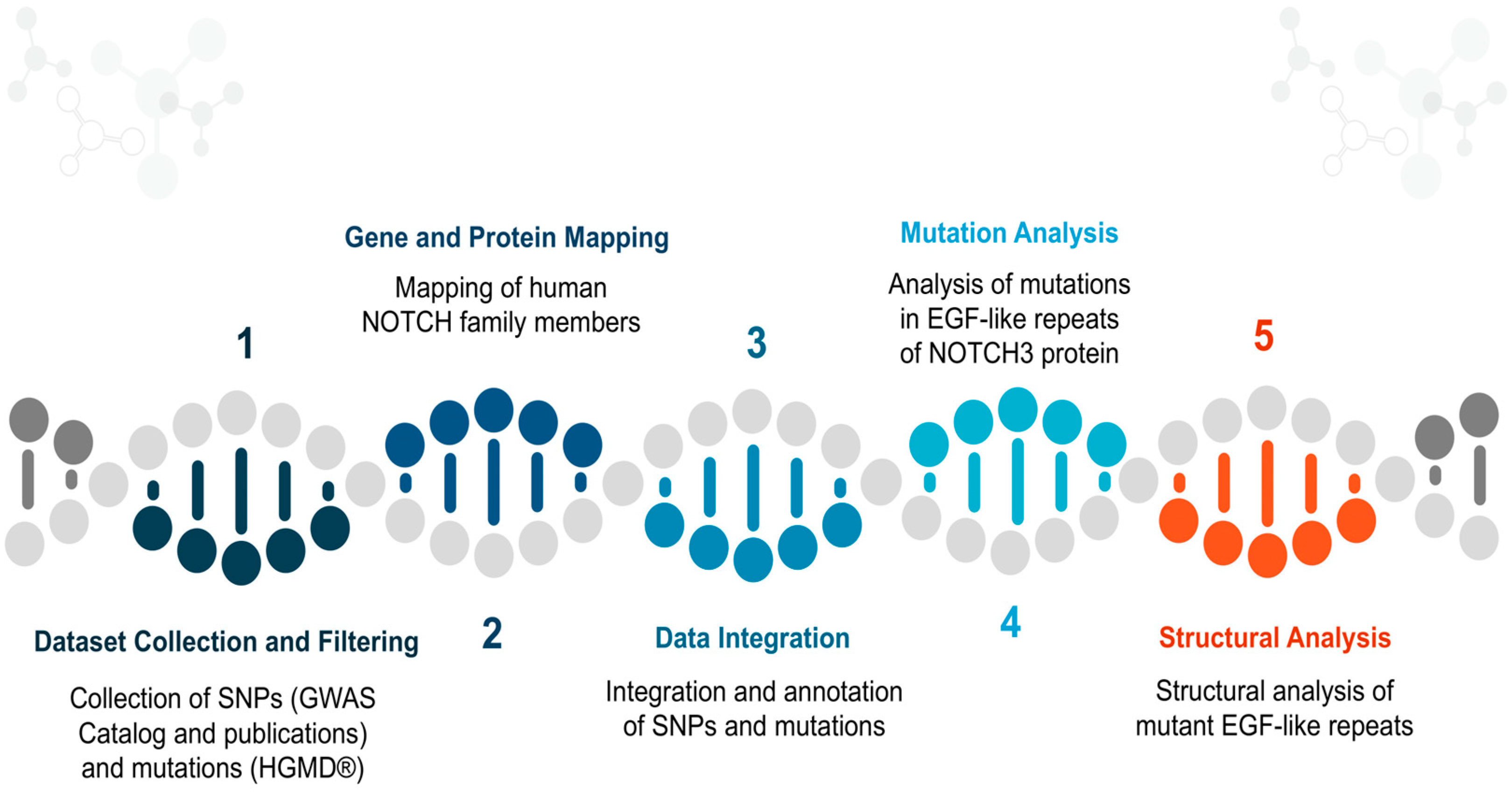
2.1. Dataset Collection and Filtering
2.2. Gene and Protein Mapping
2.3. Data Integration
2.4. Mutation Analysis
2.5. Structural Analysis
3. Results
3.1. Dataset
3.2. Gene and Protein Mapping
3.3. Data Integration
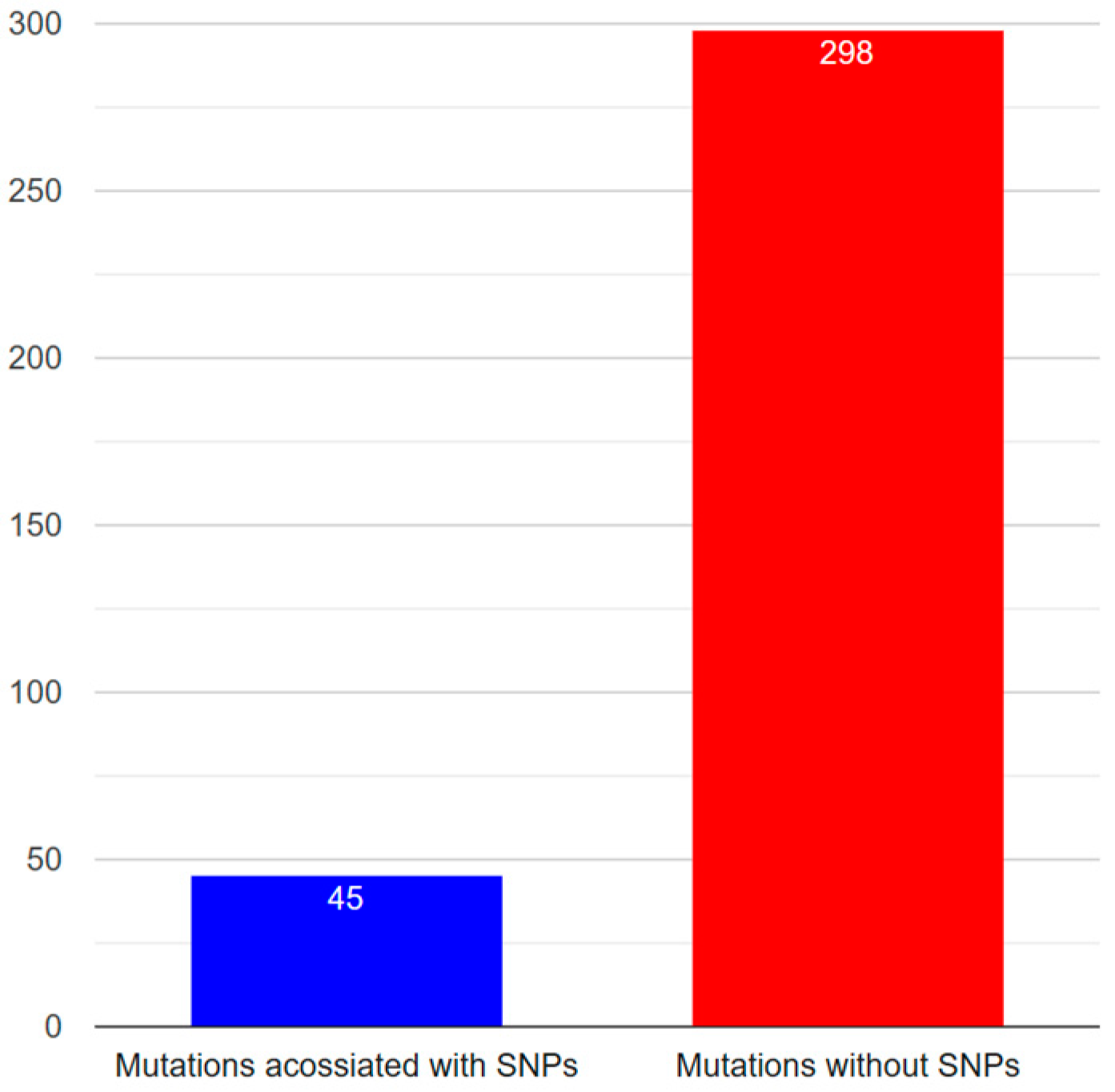
3.4. Mutation Analysis
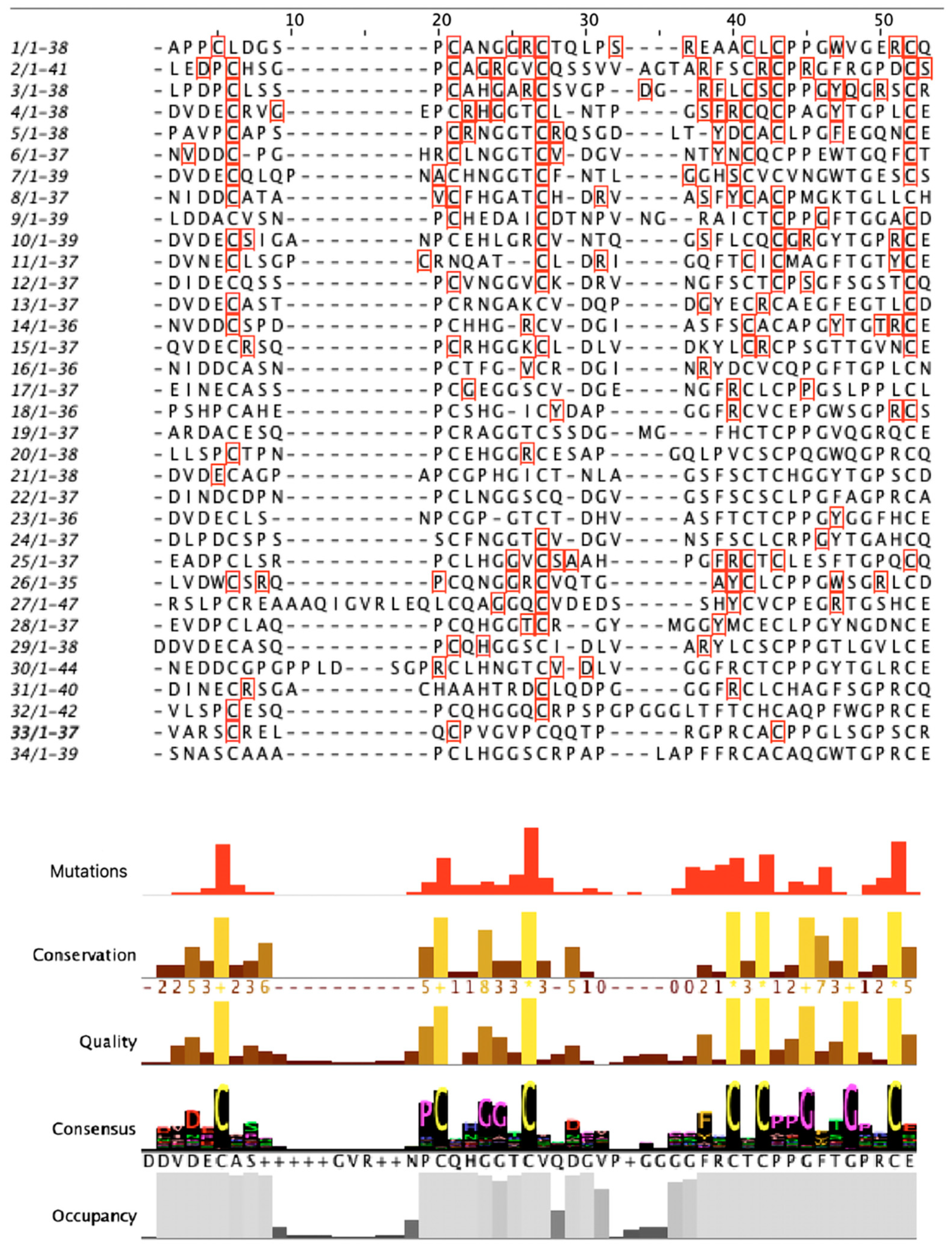
3.5. Structural Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mohr, O.L. Character Changes Caused by Mutation of an Entire Region of a Chromosome in Drosophila. Genetics 1919, 4, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Vlachakis, D.; Papageorgiou, L.; Papadaki, A.; Georga, M.; Kossida, S.; Eliopoulos, E. An updated evolutionary study of the Notch family reveals a new ancient origin and novel invariable motifs as potential pharmacological targets. PeerJ 2020, 8, e10334. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, S.; Muskavitch, M.A.; Yedvobnick, B. Molecular cloning of Notch, a locus affecting neurogenesis in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 1983, 80, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Yochem, J.; Weston, K.; Greenwald, I. The Caenorhabditis elegans lin-12 gene encodes a transmembrane protein with overall similarity to Drosophila Notch. Nature 1988, 335, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch signaling pathway: Architecture, disease, and therapeutics. Signal Transduct. Target. Ther. 2022, 7, 95. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Engler, A.; Taylor, V. Notch: An interactive player in neurogenesis and disease. Cell Tissue Res. 2018, 371, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Milner, L.A.; Bigas, A. Notch as a mediator of cell fate determination in hematopoiesis: Evidence and speculation. Blood 1999, 93, 2431–2448. [Google Scholar] [CrossRef] [PubMed]
- Dumortier, A.; Wilson, A.; MacDonald, H.R.; Radtke, F. Paradigms of notch signaling in mammals. Int. J. Hematol. 2005, 82, 277–284. [Google Scholar] [CrossRef]
- Shen, W.; Huang, J.; Wang, Y. Biological Significance of NOTCH Signaling Strength. Front. Cell Dev. Biol. 2021, 9, 652273. [Google Scholar] [CrossRef]
- Sanchez-Martin, M.; Ferrando, A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 2017, 129, 1124–1133. [Google Scholar] [CrossRef]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in NOTCH1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Xiu, M.X.; Liu, Y.M. The role of oncogenic Notch2 signaling in cancer: A novel therapeutic target. Am. J. Cancer Res. 2019, 9, 837–854. [Google Scholar] [PubMed]
- Arruga, F.; Vaisitti, T.; Deaglio, S. The NOTCH Pathway and Its Mutations in Mature B Cell Malignancies. Front. Oncol. 2018, 8, 550. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Maekawa, Y.; Kitamura, A.; Nishida, J.; Koyanagi, A.; Yagita, H.; Kojima, H.; Chiba, S.; Shimada, M.; Yasutomo, K. Notch2 signaling is required for potent antitumor immunity in vivo. J. Immunol. 2010, 184, 4673–4678. [Google Scholar] [CrossRef] [PubMed]
- Canalis, E.; Zanotti, S. Hajdu-Cheney Syndrome, a Disease Associated with NOTCH2 Mutations. Curr. Osteoporos. Rep. 2016, 14, 126–131. [Google Scholar] [CrossRef]
- Turnpenny, P.D.; Ellard, S. Alagille syndrome: Pathogenesis, diagnosis and management. Eur. J. Hum. Genet. 2012, 20, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Manini, A.; Pantoni, L. CADASIL from Bench to Bedside: Disease Models and Novel Therapeutic Approaches. Mol. Neurobiol. 2021, 58, 2558–2573. [Google Scholar] [CrossRef]
- Hosseini-Alghaderi, S.; Baron, M. Notch3 in Development, Health and Disease. Biomolecules 2020, 10, 485. [Google Scholar] [CrossRef] [PubMed]
- Snijders, A.M.; Schmidt, B.L.; Fridlyand, J.; Dekker, N.; Pinkel, D.; Jordan, R.C.; Albertson, D.G. Rare amplicons implicate frequent deregulation of cell fate specification pathways in oral squamous cell carcinoma. Oncogene 2005, 24, 4232–4242. [Google Scholar] [CrossRef]
- Xiu, M.; Zeng, X.; Shan, R.; Wen, W.; Li, J.; Wan, R. Targeting Notch4 in Cancer: Molecular Mechanisms and Therapeutic Perspectives. Cancer Manag. Res. 2021, 13, 7033–7045. [Google Scholar] [CrossRef]
- Kapoor, A.; Nation, D.A. Role of Notch signaling in neurovascular aging and Alzheimer’s disease. Semin. Cell Dev. Biol. 2021, 116, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Guerra, J.; Valadao, A.L.; Vlachakis, D.; Polak, K.; Vila, I.K.; Taffoni, C.; Prabakaran, T.; Marriott, A.S.; Kaczmarek, R.; Houel, A.; et al. Lysyl-tRNA synthetase produces diadenosine tetraphosphate to curb STING-dependent inflammation. Sci. Adv. 2020, 6, eaax3333. [Google Scholar] [CrossRef] [PubMed]
- Basak, O.; Giachino, C.; Fiorini, E.; Macdonald, H.R.; Taylor, V. Neurogenic subventricular zone stem/progenitor cells are Notch1-dependent in their active but not quiescent state. J. Neurosci. 2012, 32, 5654–5666. [Google Scholar] [CrossRef]
- Nyfeler, Y.; Kirch, R.D.; Mantei, N.; Leone, D.P.; Radtke, F.; Suter, U.; Taylor, V. Jagged1 signals in the postnatal subventricular zone are required for neural stem cell self-renewal. EMBO J. 2005, 24, 3504–3515. [Google Scholar] [CrossRef]
- Lv, Y.; Pang, X.; Cao, Z.; Song, C.; Liu, B.; Wu, W.; Pang, Q. Evolution and Function of the Notch Signaling Pathway: An Invertebrate Perspective. Int. J. Mol. Sci. 2024, 25, 3322. [Google Scholar] [CrossRef]
- Sprinzak, D.; Blacklow, S.C. Biophysics of Notch Signaling. Annu. Rev. Biophys. 2021, 50, 157–189. [Google Scholar] [CrossRef]
- Urban, N.; van den Berg, D.L.; Forget, A.; Andersen, J.; Demmers, J.A.; Hunt, C.; Ayrault, O.; Guillemot, F. Return to quiescence of mouse neural stem cells by degradation of a proactivation protein. Science 2016, 353, 292–295. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [PubMed]
- Jagaran, K.; Singh, M. Nanomedicine for Neurodegenerative Disorders: Focus on Alzheimer’s and Parkinson’s Diseases. Int. J. Mol. Sci. 2021, 22, 9082. [Google Scholar] [CrossRef] [PubMed]
- Vlachakis, D.; Tsaniras, S.C.; Ioannidou, K.; Papageorgiou, L.; Baumann, M.; Kossida, S. A series of Notch3 mutations in CADASIL; insights from 3D molecular modelling and evolutionary analyses. J. Mol. Biochem. 2014, 3, 134. [Google Scholar]
- Ables, J.L.; Breunig, J.J.; Eisch, A.J.; Rakic, P. Not(ch) just development: Notch signalling in the adult brain. Nat. Rev. Neurosci. 2011, 12, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.M. Cadasil. Handb. Clin. Neurol. 2018, 148, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Mizuta, I.; Nakao-Azuma, Y.; Yoshida, H.; Yamaguchi, M.; Mizuno, T. Progress to Clarify How NOTCH3 Mutations Lead to CADASIL, a Hereditary Cerebral Small Vessel Disease. Biomolecules 2024, 14, 127. [Google Scholar] [CrossRef] [PubMed]
- Louvi, A.; Arboleda-Velasquez, J.F.; Artavanis-Tsakonas, S. CADASIL: A critical look at a Notch disease. Dev. Neurosci. 2006, 28, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N.; Kittner, S.J. Top-NOTCH3 Variants in the Population at Large. Stroke 2020, 51, 3482–3484. [Google Scholar] [CrossRef] [PubMed]
- Haltom, A.R.; Jafar-Nejad, H. The multiple roles of epidermal growth factor repeat O-glycans in animal development. Glycobiology 2015, 25, 1027–1042. [Google Scholar] [CrossRef]
- Zeng, Q.; Pan, H.; Zhao, Y.; Wang, Y.; Xu, Q.; Tan, J.; Yan, X.; Li, J.; Tang, B.; Guo, J. Association between NOTCH3 gene and Parkinson’s disease based on whole-exome sequencing. Front. Aging Neurosci. 2022, 14, 995330. [Google Scholar] [CrossRef] [PubMed]
- Szymanowicz, O.; Korczowska-Lacka, I.; Slowikowski, B.; Wiszniewska, M.; Piotrowska, A.; Goutor, U.; Jagodzinski, P.P.; Kozubski, W.; Dorszewska, J. Headache and NOTCH3 Gene Variants in Patients with CADASIL. Neurol. Int. 2023, 15, 1238–1252. [Google Scholar] [CrossRef] [PubMed]
- Rutten, J.W.; Van Eijsden, B.J.; Duering, M.; Jouvent, E.; Opherk, C.; Pantoni, L.; Federico, A.; Dichgans, M.; Markus, H.S.; Chabriat, H.; et al. The effect of NOTCH3 pathogenic variant position on CADASIL disease severity: NOTCH3 EGFr 1-6 pathogenic variant are associated with a more severe phenotype and lower survival compared with EGFr 7-34 pathogenic variant. Genet. Med. 2019, 21, 676–682. [Google Scholar] [CrossRef]
- Matsushima, T.; Conedera, S.; Tanaka, R.; Li, Y.; Yoshino, H.; Funayama, M.; Ikeda, A.; Hosaka, Y.; Okuzumi, A.; Shimada, Y.; et al. Genotype-phenotype correlations of cysteine replacement in CADASIL. Neurobiol. Aging 2017, 50, 169.e7–169.e14. [Google Scholar] [CrossRef]
- Young, K.Z.; Lee, S.J.; Zhang, X.; Cartee, N.M.P.; Torres, M.; Keep, S.G.; Gabbireddy, S.R.; Fontana, J.L.; Qi, L.; Wang, M.M. NOTCH3 is non-enzymatically fragmented in inherited cerebral small-vessel disease. J. Biol. Chem. 2020, 295, 1960–1972. [Google Scholar] [CrossRef] [PubMed]
- Dichgans, M.; Ludwig, H.; Muller-Hocker, J.; Messerschmidt, A.; Gasser, T. Small in-frame deletions and missense mutations in CADASIL: 3D models predict misfolding of Notch3 EGF-like repeat domains. Eur. J. Hum. Genet. 2000, 8, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ren, Z.; Shi, Y.; Zhang, J. A Novel Mutation Outside of the EGFr Encoding Exons of NOTCH3 Gene in a Chinese with CADASIL. J. Stroke Cerebrovasc. Dis. Off. J. Natl. Stroke Assoc. 2020, 29, 105410. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Mizuta, I.; Watanabe-Hosomi, A.; Mukai, M.; Koizumi, T. Clinical and Genetic Aspects of CADASIL. Front. Aging Neurosci. 2020, 12, 91. [Google Scholar] [CrossRef]
- Sari, U.S.; Kisabay, A.; Batum, M.; Tarhan, S.; Dogan, N.; Coskunoglu, A.; Cam, S.; Yilmaz, H.; Selcuki, D. CADASIL with Atypical Clinical Symptoms, Magnetic Resonance Imaging, and Novel Mutations: Two Case Reports and a Review of the Literature. J. Mol. Neurosci. 2019, 68, 529–538. [Google Scholar] [CrossRef]
- Servito, M.; Gill, I.; Durbin, J.; Ghasemlou, N.; Popov, A.F.; Stephen, C.D.; El-Diasty, M. Management of Coronary Artery Disease in CADASIL Patients: Review of Current Literature. Medicina 2023, 59, 586. [Google Scholar] [CrossRef]
- Mitchell, A.; Chang, H.Y.; Daugherty, L.; Fraser, M.; Hunter, S.; Lopez, R.; McAnulla, C.; McMenamin, C.; Nuka, G.; Pesseat, S.; et al. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 2015, 43, D213–D221. [Google Scholar] [CrossRef] [PubMed]
- Chillakuri, C.R.; Sheppard, D.; Lea, S.M.; Handford, P.A. Notch receptor-ligand binding and activation: Insights from molecular studies. Semin. Cell Dev. Biol. 2012, 23, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Papakonstantinou, E.; Bacopoulou, F.; Brouzas, D.; Megalooikonomou, V.; D’Elia, D.; Bongcam-Rudloff, E.; Vlachakis, D. NOTCH3 and CADASIL syndrome: A genetic and structural overview. EMBnet J. 2019, 24, e921. [Google Scholar] [CrossRef]
- Muino, E.; Gallego-Fabrega, C.; Cullell, N.; Carrera, C.; Torres, N.; Krupinski, J.; Roquer, J.; Montaner, J.; Fernandez-Cadenas, I. Systematic Review of Cysteine-Sparing NOTCH3 Missense Mutations in Patients with Clinical Suspicion of CADASIL. Int. J. Mol. Sci. 2017, 18, 1964. [Google Scholar] [CrossRef]
- Salazar, J.L.; Yang, S.A.; Yamamoto, S. Post-Developmental Roles of Notch Signaling in the Nervous System. Biomolecules 2020, 10, 985. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.M.; Artavanis-Tsakonas, S.; Louvi, A. The Notch pathway in CNS homeostasis and neurodegeneration. Wiley Interdiscip. Rev. Dev. Biol. 2020, 9, e358. [Google Scholar] [CrossRef] [PubMed]
- Lampada, A.; Taylor, V. Notch signaling as a master regulator of adult neurogenesis. Front. Neurosci. 2023, 17, 1179011. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Zhang, X.; Wu, E.; Sukpraphrute, R.; Sukpraphrute, C.; Ye, A.; Wang, M.M. Structural changes in NOTCH3 induced by CADASIL mutations: Role of cysteine and non-cysteine alterations. J. Biol. Chem. 2023, 299, 104838. [Google Scholar] [CrossRef] [PubMed]
- Webb, E.A.; Smith, T.D.; Cotton, R.G. Difficulties in finding DNA mutations and associated phenotypic data in web resources using simple, uncomplicated search terms, and a suggested solution. Hum. Genom. 2011, 5, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Nallapareddy, V.; Bogam, S.; Devarakonda, H.; Paliwal, S.; Bandyopadhyay, D. DeepCys: Structure-based multiple cysteine function prediction method trained on deep neural network: Case study on domains of unknown functions belonging to COX2 domains. Proteins 2021, 89, 745–761. [Google Scholar] [CrossRef] [PubMed]
- Barnes, M.R. Bioinformatics for Geneticists a Bioinformatics Primer for the Analysis of Genetic Data, 2nd ed.; Wiley: Chichester, UK, 2007. [Google Scholar] [CrossRef]
- Saier, M.H.J. Understanding the Genetic Code. J. Bacteriol. 2019, 201. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S.; Maloy, S.R.; Hughes, K.T. Brenner’s encyclopedia of genetics. In Encyclopedia of Genetics, 2nd ed.; Academic Press: New York, NY, USA, 2013. [Google Scholar]
- Locatelli, M.; Padovani, A.; Pezzini, A. Pathophysiological Mechanisms and Potential Therapeutic Targets in Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL). Front. Pharmacol. 2020, 11, 321. [Google Scholar] [CrossRef] [PubMed]
- Dupe, C.; Guey, S.; Biard, L.; Dieng, S.; Lebenberg, J.; Grosset, L.; Alili, N.; Herve, D.; Tournier-Lasserve, E.; Jouvent, E.; et al. Phenotypic variability in 446 CADASIL patients: Impact of NOTCH3 gene mutation location in addition to the effects of age, sex and vascular risk factors. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow. Metab. 2023, 43, 153–166. [Google Scholar] [CrossRef]
- Oliveira, D.V.; Coupland, K.G.; Shao, W.; Jin, S.; Del Gaudio, F.; Wang, S.; Fox, R.; Rutten, J.W.; Sandin, J.; Zetterberg, H.; et al. Active immunotherapy reduces NOTCH3 deposition in brain capillaries in a CADASIL mouse model. EMBO Mol. Med. 2023, 15, e16556. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNPs | Mutations | Neurodegenerative Diseases | |
|---|---|---|---|
| Notch1 | 1 | 1 | Mental retardation, autosomal dominant, Alzheimer’s disease |
| Notch2 | 1 | 1 | Autism multiplex |
| Notch3 | 28 | 312 | CADASIL, white matter lesions, Alzheimer’s disease, ischemic stroke, cerebral small-vessel disease, arteriopathy and cavitating leukoencephalopathy, autism, migraine |
| Notch4 | 0 | 4 | Schizophrenia, multiple sclerosis, migraine |
| Gene | Notch1 | Notch2 | Notch3 | Notch4 |
|---|---|---|---|---|
| Locus | 9q34.3 | 1p12 | 19p13.12 | 6p21.32 |
| DNA linear | 58343 bp | 165110 bp | 48349 bp | 36225 bp |
| Exon count | 34 | 34 | 33 | 31 |
| Accession number | NG_007458 | NG_008163 | NG_009819 | NG_028190 |
| Organism | Homo sapiens | Homo sapiens | Homo sapiens | Homo sapiens |
| Also known as | hN1; AOS5; TAN1; AOVD1 | hN2; AGS2; HJCYS | IMF2; LMNS; CASIL; CADASIL; CADASIL1 | INT3 |
| Gene | Notch1 | Notch2 | Notch3 | Notch4 |
|---|---|---|---|---|
| Protein length | 2555 aa | 2471 aa | 2321 aa | 2003 aa |
| Accession number | NP_060087 | NP_077719 XP_946791 XP_950472 | NP_000426 | NP_004548 |
| Organism | Homo sapiens | Homo sapiens | Homo sapiens | Homo sapiens |
| Disease | T-cell acute lymphoblastic leukemia, Adams–Oliver syndrome, aortic valve disease, cancer | Hajdu–Cheney syndrome, Alagille syndrome, cancer | CADASIL, infantile myofibromatosis, early-onset arteriopathy with cavitating leukodystrophy, lateral meningocele syndrome, cancer | Schizophrenia |
| Notch1 | ||||||
|---|---|---|---|---|---|---|
| A/A | SNP ID | Nt Change NG_007458.1 | Mutation | Domain | Phenotype | Accession Number |
| 1 | _ | _ | Y2116>TERM | TAD | Mental retardation, autosomal dominant | CM171556 |
| 2 | rs79782048 | 36150G>A | Ε694>Κ | EGF-like 18 | Alzheimer’s disease | |
| Notch2 | ||||||
|---|---|---|---|---|---|---|
| A/A | SNP ID | Nt Change NG_008163.1 | Mutation | Domain | Phenotype | Accession Number |
| 1 | rs61752484 | 148130A>G | D1327G | EGF-like 34 | autism multiplex | |
| Notch3 | |||||||
|---|---|---|---|---|---|---|---|
| A/A | SNP ID | nt Change NG_009819.1 | Codon Change | Mutation | Domain | Phenotype | Accession Number |
| 1 | N/A | TGC-GGC | C43>G | EGF-like 1 | CADASIL | CM052264 | |
| 2 | N/A | TGC-TCC | C43>S | CM1712096 | |||
| 3 | N/A | TGC-TTC | C43>F | CM035648 | |||
| 4 | N/A | TGT-CGT | C49>R | CM106868 | |||
| 5 | N/A | TGT-GGT | C49>G | CM073243 | |||
| 6 | rs193921045 | 8431G>A | TGT-TAT | C49>Y | CM971054 | ||
| 7 | rs193921045 | 8431G>T | TGT-TTT | C49>F | CM052265 | ||
| 8 | N/A | GGT-TGT | G53>C | CM106869 | |||
| 9 | N/A | CGT-TGT | R54>C | CM003012 | |||
| 10 | N/A | TGC-GGC | C55>G | CM1714276 | |||
| 11 | N/A | TCC-TGC | S60>C | CM052266 | |||
| 12 | N/A | CGG-TGG | R61>W | CM0910982 | |||
| 13 | N/A | TGC-GGC | C65>G | CM108953 | |||
| 14 | N/A | TGC-TAC | C65>Y | HM070086 | |||
| 15 | N/A | TGC-TCC | C65>S | CM052267 | |||
| 16 | N/A | TGC-AGC | C67>S | CM092086 | |||
| 17 | N/A | TGC-TAC | C67>Y | CM034666 | |||
| 18 | rs28937321 | 13478G>T | TGG-TGT | W71>C | |||
| 19 | N/A | CGG-CCG | R75>Q | CM156963 | |||
| 20 | N/A | CGG-CCG | R75>P | CM061880 | |||
| 21 | N/A | TGT-CGT | C76>R | CM023649 | |||
| 22 | N/A | TGT-TGG | C76>W | CM052268 | |||
| 23 | N/A | TGT-TAT | C76>Y | CM1615079 | |||
| 24 | N/A | GAC-GGC | D80>G | EGF-like 2 | CADASIL | CM150655 | |
| 25 | N/A | TGT-CGT | C82>R | CM148255 | |||
| 26 | N/A | TGT-CGT | C87>R | CM052269 | |||
| 27 | N/A | TGT-TAT | C87>Y | CM044913 | |||
| 28 | N/A | TGT-TTT | C87>F | CM1714277 | |||
| 29 | N/A | GGC-TGC | G89>C | CM128781 | |||
| 30 | N/A | CGT-TGT | R90>C | CM971055 | |||
| 31 | N/A | TGC-GGC | C93>G | CM1714278 | |||
| 32 | N/A | TGC-TAC | C93>Y | CM023650 | |||
| 33 | N/A | TGC-TTC | C93>F | CM001263 | |||
| 34 | N/A | CGA-TGA | R103>Term | CM1313313 | |||
| 35 | N/A | TGC-CGC | C106>R | CM138997 | |||
| 36 | N/A | TGC-GGC | C106>G | CM156032 | |||
| 37 | N/A | TGC-TGG | C106>W | CM044912 | |||
| 38 | N/A | CGG-TGG | R107>W | CM159172 | |||
| 39 | N/A | TGC-AGC | C108>S | CM125146 | |||
| 40 | N/A | TGC-CGC | C108>R | CM045072 | |||
| 41 | N/A | TGC-TAC | C108>Y | CM052270 | |||
| 42 | N/A | TGC-TGG | C108>W | HM040053 | |||
| 43 | N/A | TGC-TTC | C108>F | CM1714279 | |||
| 44 | N/A | CGT-TGT | R110>C | CM971056 | |||
| 45 | N/A | TGC-CGC | C117>R | CM078103 | |||
| 46 | N/A | TGC-TAC | C117>Y | CM0910785 | |||
| 47 | N/A | TGC-TCC | C117>S | CM132414 | |||
| 48 | N/A | TGC-TTC | C117>F | CM001264 | |||
| 49 | N/A | TCC-TGC | S118>C | HM040099 | |||
| 50 | N/A | TGC-TAC | C123>Y | EGF-like 3 | CADASIL | CM003013 | |
| 51 | N/A | TGC-TTC | C123>F | CM001265 | |||
| 52 | N/A | TGT-GGT | C128>G | CM064156 | |||
| 53 | N/A | TGT-TAT | C128>Y | CM023651 | |||
| 54 | N/A | TGT-TTT | C128>F | CM125156 | |||
| 55 | N/A | GGT-TGT | G131>C | HM070153 | |||
| 56 | rs137852642 | 13740C>T | CGC-TGC | R133>C | CM971057 | ||
| 57 | rs137852642 | 13740C>A | CGC-AGC | R133>S | |||
| 58 | N/A | TGC-GGC | C134>G | CM156958 | |||
| 59 | N/A | TGC-TAC | C134>Y | CM125159 | |||
| 60 | N/A | TGC-TGG | C134>W | CM014589 | |||
| 61 | N/A | GAT-GGT | D139>V | CM1615017 | |||
| 62 | N/A | CGC-TGC | R141>C | CM971058 | |||
| 63 | N/A | TTC-TGC | F142>C | CM023652 | |||
| 64 | N/A | TGC-TCC | C144>S | CM159357 | |||
| 65 | N/A | TGC-TAC | C144>Y | CM001267 | |||
| 66 | N/A | TGC-TTC | C144>F | CM003947 | |||
| 67 | N/A | TCC-TGC | S145>C | CM044908 | |||
| 68 | N/A | TGC-CGC | C146>R | CM971059 | |||
| 69 | N/A | TGC-TAC | C146>Y | CM045307 | |||
| 70 | N/A | TGC-TGG | C146>W | CM1213543 | |||
| 71 | N/A | GGC-GTC | G149>V | CM147538 | |||
| 72 | N/A | GGC-TGC | G149>C | CM052271 | |||
| 73 | N/A | TAC-TGC | Y150>C | CM001268 | |||
| 74 | rs371491165 | 13794C>G | CAG-GAG | Q151>E | CM0910786 | ||
| 75 | rs797045014 | 13800C>T | CGC-TGC | R153>C | CM971060 | ||
| 76 | rs797045014 | 13800C>A | CGC-AGC | R153>S | |||
| 77 | N/A | TGC-AGC | C155>S | CM0910788 | |||
| 78 | N/A | TGC-TAC | C155>Y | CM125157 | |||
| 79 | N/A | TGC-TCC | C155>S | CM044910 | |||
| 80 | N/A | TGC-TGG | C155>W | CM159358 | |||
| 81 | N/A | TGC-AGC | C162>S | EGF-like 4 | CADASIL | CM003014 | |
| 82 | N/A | TGC-CGC | C162>R | CM086704 | |||
| 83 | N/A | TGC-TAC | C162>Y | CM170225 | |||
| 84 | N/A | TGC-TGG | C162>W | CM035650 | |||
| 85 | N/A | GGT-TGT | G165>C | CM0910787 | |||
| 86 | rs28933696 | 13848C>T | CGC-TGC | R169>C | CM961043 | ||
| 87 | rs147373451 | 13852A>G | CAT-CGT | H170>R | CADASIL, Alzheimer’s disease | CM107598 | |
| 88 | rs147373451 | 13852A>T | CAT-CTT | H170>L | CADASIL, Alzheimer’s disease | ||
| 89 | N/A | GGT-TGT | G171>C | CM971061 | |||
| 90 | N/A | TGC-AGC | C174>S | CM125160 | |||
| 91 | N/A | TGC-CGC | C174>R | CM033795 | |||
| 92 | N/A | TGC-TAC | C174>Y | CM001269 | |||
| 93 | N/A | TGC-TTC | C174>F | CM014211 | |||
| 94 | N/A | TCC-TGC | S180>C | CM003015 | |||
| 95 | N/A | TTC-TGC | F181>C | CM095734 | |||
| 96 | rs28933697 | 13887C>T | CGC-TGC | R182>C | CADASIL, Alzheimer’s disease | CM961044 | |
| 97 | N/A | TGC-AGC | C183>S | CM001271 | |||
| 98 | N/A | TGC-CGC | C183>R | CM001270 | |||
| 99 | N/A | TGC-TAC | C183>Y | CM1615080 | |||
| 100 | N/A | TGC-TTC | C183>F | CM052272 | |||
| 101 | N/A | TGT-AGT | C185>S | CM1010137 | |||
| 102 | N/A | TGT-CGT | C185>R | CM971062 | |||
| 103 | N/A | TGT-GGT | C185>G | CM014590 | |||
| 104 | N/A | TGT-TAT | C185>Y | CM147761 | |||
| 105 | N/A | TAC-TGC | Y189>C | CM042442 | |||
| 106 | N/A | TGT-AGT | C194>S | CM042443 | |||
| 107 | N/A | TGT-CGT | C194>R | CM023653 | |||
| 108 | N/A | TGT-GGT | C194>G | CM150660 | |||
| 109 | N/A | TGT-TCT | C194>S | CM1010138 | |||
| 110 | N/A | TGT-TTT | C194>F | CM001272 | |||
| 111 | N/A | TGT-TAT | C194>Y | CM003016 | |||
| 112 | N/A | GCG-ACG | A198>T | White matter lesions? | CM0911492 | ||
| 113 | N/A | TGT-CGT | C201>R | CM065340 | |||
| 114 | N/A | TGT-TAT | C201>Y | CM044914 | |||
| 115 | N/A | GCG-GTG | A201>V | CADASIL | CM121679 | ||
| 116 | N/A | TGC-CGC | C206>R | EGF-like 5 | CADASIL | CM055455 | |
| 117 | N/A | TGC-TAC | C206>Y | CM003017 | |||
| 118 | N/A | CGT-CAT | R207>H | White matter lesions? | CM0911493 | ||
| 119 | N/A | CGT-TGT | R207>C | CADASIL | CM003018 | ||
| 120 | N/A | TGC-AGC | C212>S | CM971063 | |||
| 121 | N/A | TGC-CGC | C212>R | CM1714280 | |||
| 122 | N/A | TGC-TAC | C212>Y | CM110280 | |||
| 123 | N/A | TGC-TGG | C212>W | CM132412 | |||
| 124 | N/A | TGC-TTC | C212>F | CM1515586 | |||
| 125 | N/A | AGG-AAG | R213>K | CM033796 | |||
| 126 | N/A | TAC-TGC | Y220>C | HM0657 | |||
| 127 | N/A | TGT-AGT | C222>S | BM1486714 | |||
| 128 | N/A | TGT-CGT | C222>R | BM1496778 | |||
| 129 | N/A | TGT-GGT | C222>G | CM971064 | |||
| 130 | N/A | TGT-TAT | C222>Y | CM023654 | |||
| 131 | N/A | TGT-TCT | C222>S | CM106870 | |||
| 132 | N/A | TGT-CGT | C224>R | CM159178 | |||
| 133 | N/A | TGT-TAT | C224>Y | CM971065 | |||
| 134 | N/A | TTT-TGT | F228>C | CM1414770 | |||
| 135 | N/A | TGT-AGT | C233>S | CM023655 | |||
| 136 | N/A | TGT-CGT | C233>R | CM1010139 | |||
| 137 | N/A | TGT-TAT | C233>Y | CM052273 | |||
| 138 | N/A | TGT-TGG | C233>W | CM035651 | |||
| 139 | N/A | GTG-ATG | V237>M | EGF-like 6 | CADASIL | CM025913 | |
| 140 | N/A | TGT-TCT | C240>S | CM052274 | |||
| 141 | N/A | TGT-AGT | C245>S | CM056015 | |||
| 142 | N/A | TGT-CGT | C245>R | CM052275 | |||
| 143 | N/A | TGT-TAT | C245>Y | CM1213544 | |||
| 144 | N/A | TGC-AGC | C251>S | CM035643 | |||
| 145 | N/A | TGC-CGC | C251>R | CM023656 | |||
| 146 | N/A | TGC-GGC | C251>G | CM077212 | |||
| 147 | N/A | TGC-TAC | C251>Y | CM092087 | |||
| 148 | N/A | GTG-ATG | V252>M | CM150661 | |||
| 149 | N/A | TAT-TGT | Y258>C | CM971066 | |||
| 150 | N/A | TGC-CGC | C260>R | CM1414771 | |||
| 151 | N/A | TGC-GGC | C260>G | EGF-like 7 | CADASIL | CM095351 | |
| 152 | N/A | TGC-TAC | C260>Y | CM052276 | |||
| 153 | N/A | TGC-TTC | C260>F | CM1213545 | |||
| 154 | N/A | TGC-TTC | C271>F | HM060011 | |||
| 155 | N/A | TGT-CGT | C278>R | CADASIL | CM155988 | ||
| 156 | N/A | TGC-TAC | C291>Y | CADASIL | CM156956 | ||
| 157 | N/A | GGT-TGT | G296>C | CADASIL | CM108668 | ||
| 158 | N/A | AGC-TGC | S299>C | CADASIL | CM046101 | ||
| 159 | N/A | AGA-AAG | G309>K | White matter lesions? | CM0911494 | ||
| 160 | N/A | TGC-AGC | C311>S | CADASIL | CM1414772 | ||
| 161 | N/A | TGT-TTT | C318>F | EGF-like 8 | CADASIL | CM1615081 | |
| 162 | N/A | GTG-ATG | V322>M | CM150662 | |||
| 163 | N/A | TGC-AGC | C323>S | CM159171 | |||
| 164 | N/A | TGC-TGG | C323>W | CM167812 | |||
| 165 | N/A | TGC-TAC | C329>Y | CM1213546 | |||
| 166 | rs137852641 | 14516C>T | CGC-TGC | R332>C | CM014070 | ||
| 167 | N/A | TCT-TGT | S335>C | CM052277 | |||
| 168 | N/A | TAC-TGC | Y337>C | CM052278 | |||
| 169 | N/A | TGT-CGT | C338>R | CM056018 | |||
| 170 | N/A | TGT-TCT | C338>S | CM1718752 | |||
| 171 | N/A | TGT-TTT | C338>F | CM150663 | |||
| 172 | N/A | TGC-TAC | C340>Y | CM1413344 | |||
| 173 | N/A | TGC-TTC | C340>F | CM1615082 | |||
| 174 | N/A | TGC-CGC | C360>R | EGF-like 9 | CADASIL | CM140149 | |
| 175 | N/A | TGT-CGT | C366>R | CM139199 | |||
| 176 | N/A | TGT-TGG | C366>W | CM082988 | |||
| 177 | N/A | TGT-CGT | C379>R | HM090054 | |||
| 178 | N/A | TGT-TCT | C379>S | CM052279 | |||
| 179 | N/A | GGC-TGC | G382>C | CM035644 | |||
| 180 | N/A | TGT-TAT | C388>Y | CM064155 | |||
| 181 | N/A | TGC-CGC | C395>R | EGF-like 10 | CADASIL | CM052280 | |
| 182 | rs863225297 | 16704C>G | TCT-TGT | S396>C | CM125147 | ||
| 183 | N/A | TGC-TAC | C408>Y | CM159179 | |||
| 184 | N/A | TCC-TGC | S414>C | CM140150 | |||
| 185 | N/A | TGC-TGG | C419>W | CM159180 | |||
| 186 | N/A | GGT-TGT | G420>C | CM023657 | |||
| 187 | N/A | CGT-TGT | R421>C | CM052281 | |||
| 188 | N/A | CGC-TGC | R427>C | CM108354 | |||
| 190 | N/A | TGT-CGT | C428>R | CM056019 | |||
| 191 | N/A | TGT-TAT | C428>Y | CM052282 | |||
| 192 | rs267606915 | 16897T>A | TGT-TCT | C428>S | CM014591 | ||
| 193 | N/A | TGT-CGT | C435>R | EGF-like 11 | CADASIL | CM035646 | |
| 194 | N/A | TGC-AGC | C440>S | CM056017 | |||
| 195 | N/A | TGC-CGC | C440>R | CM052283 | |||
| 196 | N/A | TGC-GGC | C440>G | CM023658 | |||
| 197 | N/A | TGC-GGC | C446>G | CM1414773 | |||
| 198 | N/A | TGC-TCC | C446>S | CM044911 | |||
| 199 | N/A | TGC-TTC | C446>F | CM035649 | |||
| 200 | N/A | CGC-TGC | R449>C | CM023659 | |||
| 201 | rs28933698 | 16978T>A | TGT-AGT | C455>S | |||
| 202 | rs28933698 | 16978T>C | TGT-CGT | C455>R | CM021648 | ||
| 203 | N/A | TGT-TAT | C455>Y | CM140151 | |||
| 204 | N/A | TGT-TCT | C457>S | CM1010140 | |||
| 205 | N/A | TAT-TGT | Y465>C | CM035647 | |||
| 206 | N/A | TGC-TAC | C466>Y | CM1111832 | |||
| 207 | N/A | TGC-TAT | C478>Y | EGF-like 12 | CADASIL | CM150326 | |
| 208 | N/A | TGC-GGC | C484>G | CADASIL | CM125158 | ||
| 209 | N/A | TGC-TAC | C484>Y | CADASIL | CM044909 | ||
| 210 | N/A | TGC-TTC | C484>F | CADASIL | CM052284 | ||
| 211 | N/A | TGC-TAC | C495>Y | CADASIL | CM052285 | ||
| 212 | rs11670799 | 17742C>T | CCC-CTC | P496>L | Ischemic stroke, Alzheimer’s disease, cerebral small-vessel disease | ||
| 213 | rs114207045 | 17745C>G | TCG-TGG | S497>W | CADASIL, white matter lesions ? | ||
| 214 | rs114207045 | 17745C>T | TCG-TTG | S497>L | CADASIL, white matter lesions ? | CM119547 | |
| 215 | N/A | TGT-CGT | C504>R | CADASIL | CM0911339 | ||
| 216 | N/A | TGC-CGC | C511>R | CADASIL | CM052286 | ||
| 217 | N/A | TGC-TAC | C511>Y | CADASIL | HM050011 | ||
| 218 | N/A | TGC-TTC | C511>F | CADASIL | CM115174 | ||
| 219 | N/A | GGC-TGC | G528>C | CADASIL | CM056020 | ||
| 220 | N/A | TGC-GGC | C531>G | Leukoencephalopathy, vascular | CM175386 | ||
| 221 | N/A | TGC-TCC | C531>S | CADASIL | HM0684 | ||
| 222 | N/A | CGC-TGC | R532>C | EGF-like 13 | CADASIL | HM070085 | |
| 223 | N/A | TGT-CGT | C542>R | CM140152 | |||
| 224 | N/A | TGT-TAT | C542>Y | CM961045 | |||
| 225 | N/A | CGC-TGC | R544>C | CM994179 | |||
| 226 | N/A | TGC-CGC | C549>R | CM035645 | |||
| 227 | N/A | TGC-TAC | C549>Y | CM052287 | |||
| 228 | N/A | CGC-TGC | R558>C | CM961046 | |||
| 229 | N/A | TGT-TAT | C568>Y | HM0710 | |||
| 230 | N/A | TAC-TGC | Y574>C | EGF-like 14 | CADASIL | HM0685 | |
| 231 | N/A | ACA-GCA | T577>A | HM0718 | |||
| 232 | N/A | CGC-TGC | R578>C | CM961047 | |||
| 233 | N/A | TGC-CGC | C579>R | CM121680 | |||
| 234 | N/A | CGC-TGC | R587>C | EGF-like 15 | CADASIL | CM061879 | |
| 235 | N/A | TGC-CGC | C591>R | CM125164 | |||
| 236 | N/A | CGC-AGC | R592>S | White matter lesions? | CM0911495 | ||
| 237 | N/A | CGC-TGC | R592>C | CADASIL | CM107182 | ||
| 238 | N/A | TGC-TAC | C597>Y | CM1615083 | |||
| 239 | N/I | TGC-TCC | C597>S | CM119361 | |||
| 240 | N/A | TGC-TGG | C597>W | CM1615018 | |||
| 241 | N/A | TGC-CGC | C606>R | CM125148 | |||
| 242 | N/A | CGC-CAC | R607>H | CM1615019 | |||
| 243 | N/A | CGC-TGC | R607>C | CM003019 | |||
| 244 | N/A | TGC-TGG | C617>W | CM1610629 | |||
| 245 | N/A | GTC-GTT | V633>V | EGF-like 16 | CADASIL | CM124698 | |
| 246 | N/A | CGC-TGC | R640>C | CADASIL | CM125168 | ||
| 247 | N/A | GTC-GAC | V644>D | White matter lesions? | CM0911496 | ||
| 248 | N/A | GGC-TGC | G667>C | EGF-like 17 | CADASIL | CM125169 | |
| 249 | rs10406745 | 20390G>C | CGC-CCC | R680>P | CADASIL | ||
| 250 | rs10406745 | 20390G>A | CGC-CAC | R680>H | CADASIL | ||
| 251 | N/A | CGC-TGC | R680>C | CADASIL with intracerebral haemorrhage? | CM122007 | ||
| 252 | N/A | CCT-ACT | P685>T | CADASIL | CM111340 | ||
| 253 | N/A | TAT-TGT | Y710>C | EGF-like 18 | CADASIL | CM1313312 | |
| 254 | N/A | CGC-TGC | R717>C | CM1414856 | |||
| 255 | N/A | CGC-TGC | R728>C | CM971067 | |||
| 256 | N/A | TGC-GGC | C729>G | CM1714281 | |||
| 257 | N/A | GTC-GCC | V764>A | EGF-like 19 | White matter lesions? | CM119548 | |
| 258 | N/A | TGC-TCC | C775>S | EGF-like 20 | CADASIL | CM052288 | |
| 259 | N/A | CGC-TGC | R785>C | CADASIL | CM1413345 | ||
| 260 | N/A | GAG-GAA | E813>E | EGF-like 21 | CADASIL | CM1615020 | |
| 261 | N/A | TAC-TGC | Y916>C | EGF-like 23 | CADASIL | CM1414857 | |
| 262 | N/A | TGT-CGT | C939>R | EGF-like 24 | CADASIL | CM125149 | |
| 263 | N/A | GGC-TGC | G953>C | EGF-like 24 | CADASIL | CM023660 | |
| 264 | N/A | TGC-TGA | C966>TERM | EGF-like 25 | Arteriopathy and cavitating leukoencephalopathy | CM152731 | |
| 265 | N/A | GGC-TGC | G975>C | CADASIL with intracerebral haemorrhage | CM067439 | ||
| 266 | N/A | TGC-AGC | C977>S | CADASIL | HM050017 | ||
| 267 | N/A | TGC-GGC | C977>G | CM1615021 | |||
| 268 | N/A | AGC-CGC | S978>R | HM0711 | |||
| 269 | N/A | GCC-TCC | A979>S | CM1313735 | |||
| 270 | N/A | TTC-TGC | F984>C | CM003020 | |||
| 271 | N/A | CGC-TGC | R985>C | CM971068 | |||
| 272 | N/A | TGC-CGC | C986>R | CM1414774 | |||
| 273 | N/A | TGC-TAC | C988>Y | CM062927 | |||
| 274 | N/A | TGC-TTC | C988>F | CM1714282 | |||
| 275 | N/A | TGC-GGC | C997>G | HM070152 | |||
| 276 | N/A | TGC-GGC | C1004>G | EGF-like 26 | CADASIL CADASIL | CM1714283 | |
| 277 | N/A | TGC-TAC | C1004>Y | EGF-like 26 | CM082987 | ||
| 278 | N/A | CGC-TGC | R1006>C | CM971069 | |||
| 279 | N/A | CCT-TCT | P1008>S | CM148526 | |||
| 280 | N/A | GGT-TGT | G1013>C | CM125150 | |||
| 281 | N/A | TGC-AGC | C1015>S | CM156030 | |||
| 282 | N/A | TGC-CGC | C1015>R | CM994180 | |||
| 283 | rs35769976 | 25217G>T | GCC-TCC | A1020>S | |||
| 284 | rs35769976 | 25217G>A | GCC-ACC | A1020>T | |||
| 285 | rs35769976 | 25217G>C | GCC-CCC | A1020>P | CM085589 | ||
| 286 | N/A | TAT-TGT | Y1021>C | CM023661 | |||
| 287 | N/A | TGC-TTC | C1022>F | CM118356 | |||
| 288 | N/A | TGG-TCG | W1028>C | CM078549 | |||
| 289 | N/A | CGC-TGC | R1031>C | CM971070 | |||
| 290 | N/A | GGT-TGT | G1058>C | EGF-like 27 | CADASIL | CM014592 | |
| 291 | N/A | TGT-TAT | C1061>Y | CM127977 | |||
| 292 | N/A | TAC-TGC | Y1069>C | CM092088 | |||
| 293 | N/A | CGT-TGT | R1076>C | CM056016 | |||
| 294 | N/A | ACC-TCC | T1098>S | EGF-like 28 | CADASIL | CM106871 | |
| 295 | N/A | TGC-TAC | C1099>Y | HM0709 | |||
| 296 | N/A | TAC-TGC | Y1106>C | CM1615022 | |||
| 297 | N/A | ATG-GTG | M1107>V | Autism spectrum disorder? | CM187124 | ||
| 298 | N/A | TGC-TGG | C1131>W | EGF-like 29 | CADASIL | CM081358 | |
| 299 | rs112197217 | 26557C>G | CAC-CAG | H1133>Q | CADASIL | ||
| 300 | rs112197217 | 26557C>A | CAC-CAA | H1133>Q | Alzheimer’s disease, ischemic stroke, CADASIL | ||
| 301 | rs112197217 | 26557C>T | CAC-CAT | H1133>H | CADASIL | ||
| 302 | N/A | GGG-CGG | G1134>R | AUTISM - | CM124589 | ||
| 303 | N/A | CGC-TGC | R1143>C | CADASIL | CM1715079 | ||
| 304 | N/A | CGG-TGG | R1175>W | EGF-like 30 | CADASIL - | CM159359 | |
| 305 | rs10408676 | 26786G>A | GTG-ATG | V1183>M | Alzheimer disease, modifier of? | CM186058 | |
| 306 | N/A | GAC-GAG | D1184>E | CADASIL | CM159173 | ||
| 307 | N/A | CGC-TGC | R1210>C | CM1615084 | |||
| 308 | rs199638166 | 26903T>G | TGC-GGC | C1222>G | EGF-like 31 | CADASIL | CM1414775 |
| 309 | rs201680145 | 26930C>T | CGT-TGT | R1231>C | CADASIL | CM971071 | |
| 310 | rs201680145 | 26930C>A | CGT-AGT | R1231>S | Alzheimer’s disease | ||
| 311 | N/A | CAT-CTT | H1235>L | White matter lesions? | CM0911497 | ||
| 312 | N/A | TGC-TGG | C1250>W | EGF-like 32 | CADASIL | HM090055 | |
| 313 | N/A | TGC-CGC | C1261>R | CADASIL | CM961048 | ||
| 314 | N/A | TGC-TAC | C1261>Y | CADASIL | CM052289 | ||
| 315 | N/A | CGT-CTT | R1262>L | White matter lesions? | CM119549 | ||
| 316 | N/A | TGC-TGG | C1293>W | EGF-like 33 | CADASIL | CM186164 | |
| 317 | N/A | TGC-TTC | C1298>F | CADASIL with haemorrhagic strokes | CM135094 | ||
| 318 | N/A | TGC-TAC | C1315>Y | CADASIL | CM116899 | ||
| 319 | N/A | GCC-ACC | A1450>T | LNR2 | White matter lesions ? | CM119550 | |
| 320 | N/A | CTC-CCC | L1515>P | NOD | CADASIL | CM081357 | |
| 321 | rs141320511 | 31730C>A | CTG-ATG | L1518>M | NOD | White matter lesions? | CM119551 |
| 322 | rs141320511 | 31730C>T | CTG-TTG | L1518>L | NOD | White matter lesions, CADASIL, vascular dementia | |
| 323 | rs367543285 | 31734T>C | CTG-CCG | L1519>P | NOD | Migraine and white matter lesions | |
| 324 | N/A | CCA-CCG | P1521>P | NOD | Alzheimer disease, modifier of? | CM186056 | |
| 325 | rs78501403 | 31857G>C | CGG-CCG | R1560>P | NOD | Ischemic stroke, cerebral small-vessel disease | |
| 326 | rs78501403 | 31857G>A | CGG-CAG | R1560>Q | NOD | Ischemic stroke, cerebral small-vessel disease | |
| 327 | N/A | GGT-GAT | G1710>D | RAM | White matter lesions? | CM119552 | |
| 328 | N/A | GTG-ATG | V1762>M | RAM | CADASIL | CM1212986 | |
| 329 | N/A | GAC-AAC | D1823>N | NLS | White matter lesions? | CM119553 | |
| 330 | rs115582213 | 43458G>A | GTG-ATG | V1952>M | ANK | Alzheimer disease (modifier of?) | CM186057 |
| 331 | rs142007575 | 44385G>A | GTA-ATA | V2011>1 | NLS | Ischemic stroke | |
| 332 | rs114447350 | 44575C>T | CCG-CTG | P2074>L | unknown | Alzheimer’s disease | |
| 333 | rs114447350 | 44575C>A | CCG-CAG | P2074>Q | unknown | Alzheimer’s disease | |
| 334 | rs1044009 | 45022C>G | GCG-GGG | A2223>G | PEST | Alzheimer’s disease, CADASIL | |
| 335 | rs1044009 | 45022C>T | GCG-GTG | A2223>V | PEST | Alzheimer’s disease, CADASIL | |
| Notch4 | ||||
|---|---|---|---|---|
| A/A | Mutation | Domain | Phenotype | Accession Number |
| 1 | EGF-like 6 | Schizophrenia | CM099076 | EGF-like 6 |
| 2 | EGF-like 8 | Multiple sclerosis | CM133099 | EGF-like 8 |
| 3 | EGF-like 21 | Migraine severity | CM134239 | EGF-like 21 |
| 4 | NOD | Migraine duration | CM134237 | NOD |
| A/A | Amino Acid | Frequency | Codons | |
|---|---|---|---|---|
| 1 | Position 6 | C | Given | TGT, TGC |
| 2 | R | 8 | CGT, CGC | |
| 3 | Y | 6 | TAT, TAC | |
| 4 | F | 3 | TTT, TTC | |
| 5 | S | 3 | AGT TCT, AGC, TCC | |
| 6 | W | 3 | TGG | |
| 7 | G | 0 | GGT, GGC | |
| 1 | Position 21 | C | Given | TGT, TGC |
| 2 | R | 6 | CGT, CGC | |
| 3 | Y | 6 | TAT, TAC | |
| 4 | F | 4 | TTT, TTC | |
| 5 | S | 2 | AGT TCT, AGC, TCC | |
| 6 | W | 2 | TGG | |
| 7 | G | 2 | GGT, GGC | |
| 1 | Position 27 | C | Given | TGT, TGC |
| 2 | R | 7 | CGT, CGC | |
| 3 | Y | 13 | TAT, TAC | |
| 4 | F | 5 | TTT, TTC | |
| 5 | S | 7 | AGT TCT, AGC, TCC | |
| 6 | W | 4 | TGG | |
| 7 | G | 8 | GGT, GGC | |
| 1 | Position 41 | C | Given | TGT, TGC |
| 2 | R | 8 | CGT, CGC | |
| 3 | Y | 7 | TAT, TAC | |
| 4 | F | 5 | TTT, TTC | |
| 5 | S | 6 | AGT TCT, AGC, TCC | |
| 6 | W | 1 | TGG | |
| 7 | G | 4 | GGT, GGC | |
| 1 | Position 43 | C | Given | TGT, TGC |
| 2 | R | 5 | CGT, CGC | |
| 3 | Y | 9 | TAT, TAC | |
| 4 | F | 3 | TTT, TTC | |
| 5 | S | 5 | AGT TCT, AGC, TCC | |
| 6 | W | 3 | TGG | |
| 7 | G | 1 | GGT, GGC | |
| 1 | Position 52 | C | Given | TGT, TGC |
| 2 | R | 8 | CGT, CGC | |
| 3 | Y | 9 | TAT, TAC | |
| 4 | F | 3 | TTT, TTC | |
| 5 | S | 6 | AGT TCT, AGC, TCC | |
| 6 | W | 4 | TGG | |
| 7 | G | 3 | GGT, GGC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papageorgiou, L.; Papa, L.; Papakonstantinou, E.; Mataragka, A.; Dragoumani, K.; Chaniotis, D.; Beloukas, A.; Iliopoulos, C.; Bongcam-Rudloff, E.; Chrousos, G.P.; et al. SNP and Structural Study of the Notch Superfamily Provides Insights and Novel Pharmacological Targets against the CADASIL Syndrome and Neurodegenerative Diseases. Genes 2024, 15, 529. https://doi.org/10.3390/genes15050529
Papageorgiou L, Papa L, Papakonstantinou E, Mataragka A, Dragoumani K, Chaniotis D, Beloukas A, Iliopoulos C, Bongcam-Rudloff E, Chrousos GP, et al. SNP and Structural Study of the Notch Superfamily Provides Insights and Novel Pharmacological Targets against the CADASIL Syndrome and Neurodegenerative Diseases. Genes. 2024; 15(5):529. https://doi.org/10.3390/genes15050529
Chicago/Turabian StylePapageorgiou, Louis, Lefteria Papa, Eleni Papakonstantinou, Antonia Mataragka, Konstantina Dragoumani, Dimitrios Chaniotis, Apostolos Beloukas, Costas Iliopoulos, Erik Bongcam-Rudloff, George P. Chrousos, and et al. 2024. "SNP and Structural Study of the Notch Superfamily Provides Insights and Novel Pharmacological Targets against the CADASIL Syndrome and Neurodegenerative Diseases" Genes 15, no. 5: 529. https://doi.org/10.3390/genes15050529