
Characterization of a New Variant in ARHGAP31 Probably Involved in Adams–Oliver Syndrome in a Family with a Variable Phenotypic Spectrum
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. DNA Purification
2.3. Clinical Exome Sequencing (CES)
2.4. Sanger Sequencing of Specific Region
2.5. Array-Based Comparative Genomic Hybridization (aCGH) Profiling
2.6. RNA Purification
2.7. Gene Expression Analysis
2.8. Bioinformatic Analysis
3. Results
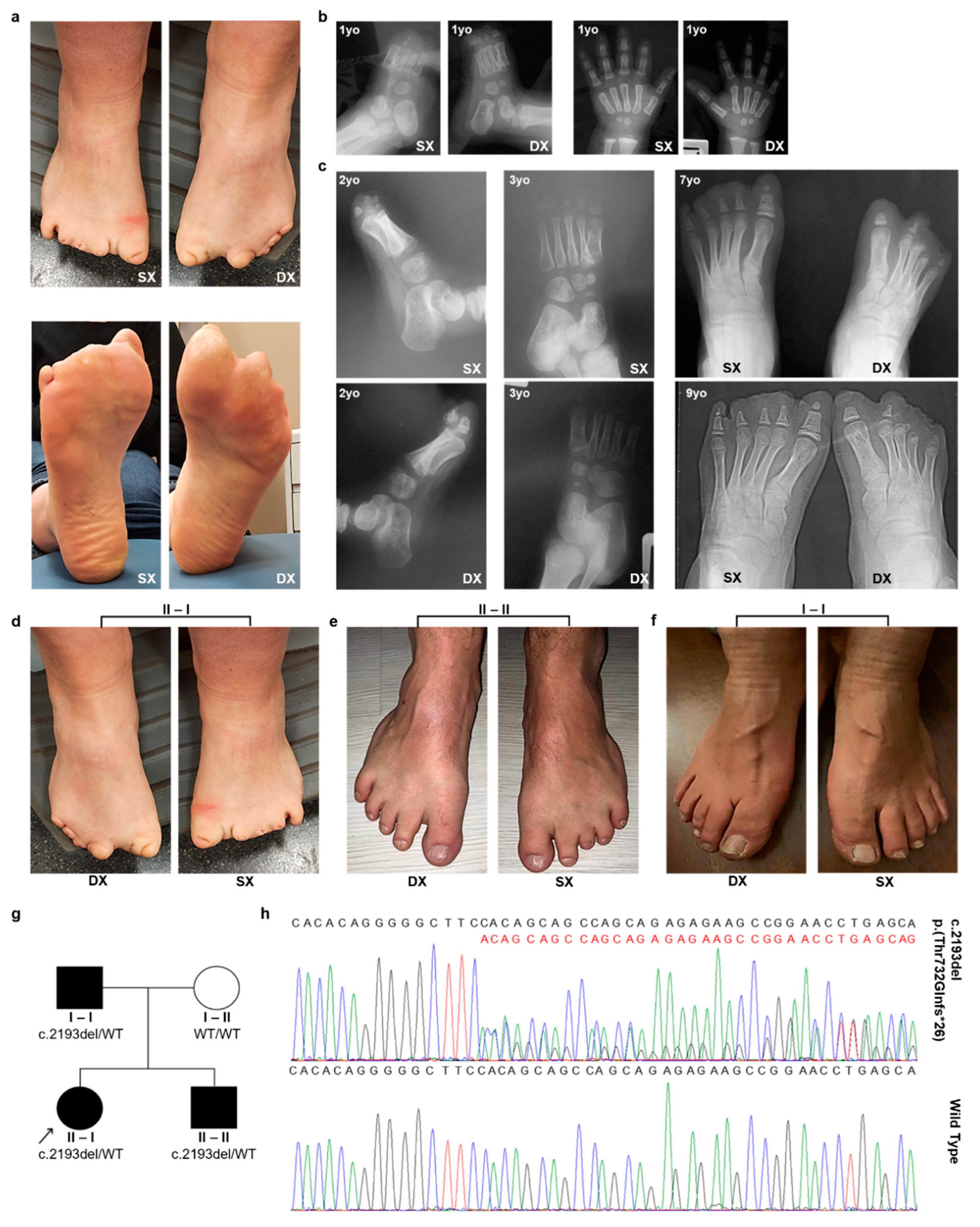
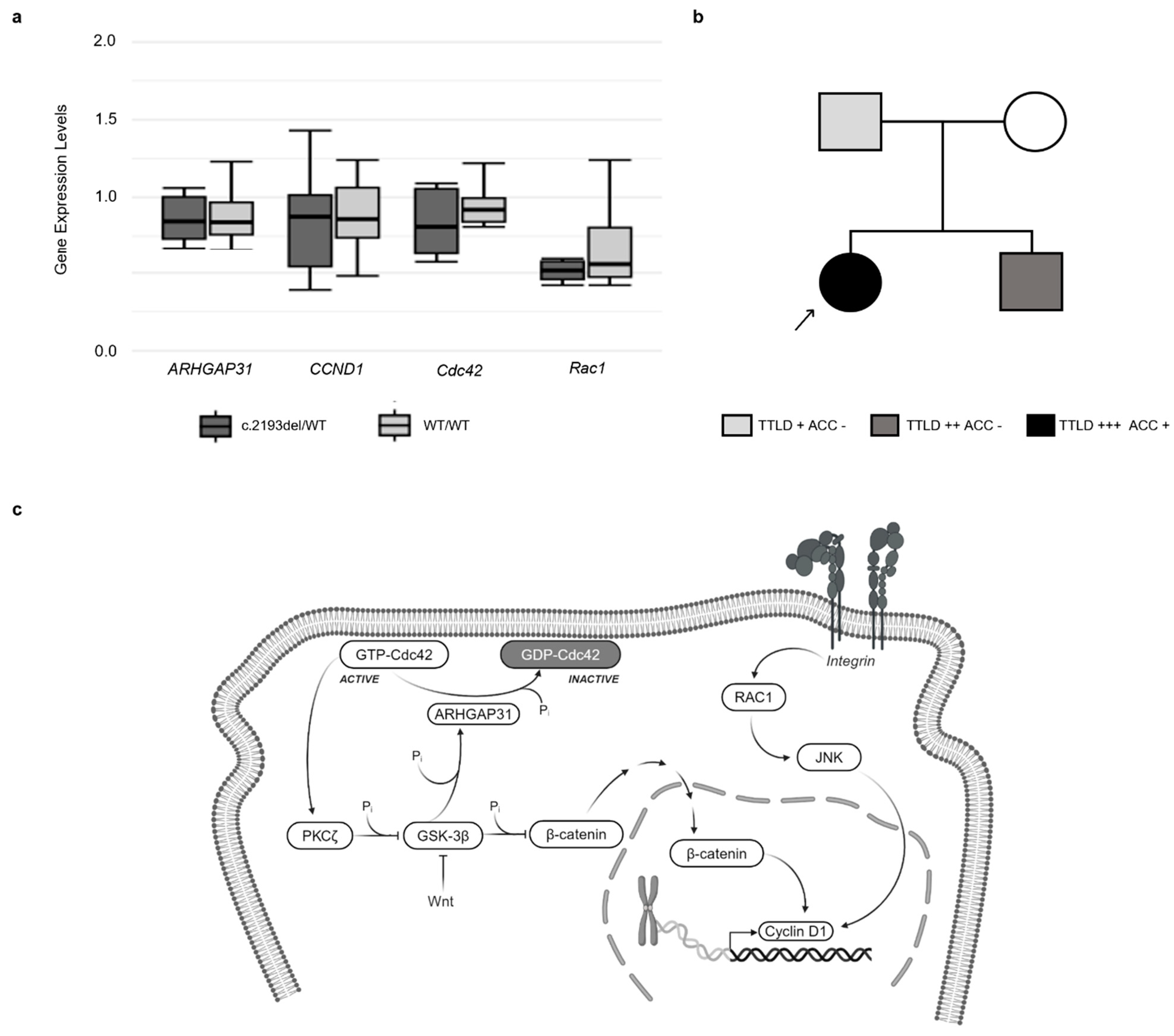
3.1. Clinical Information
3.2. Clinical Exome Analysis
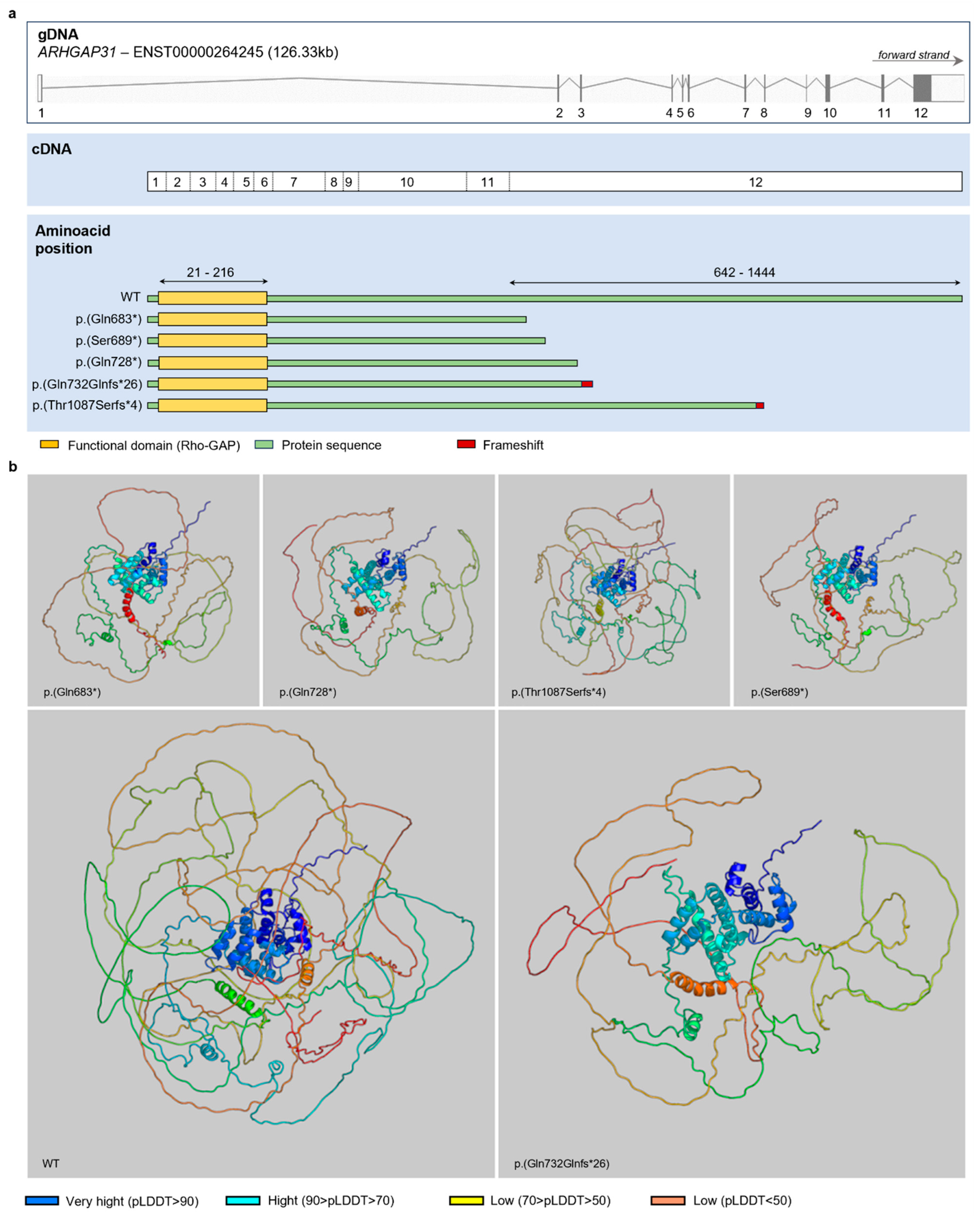
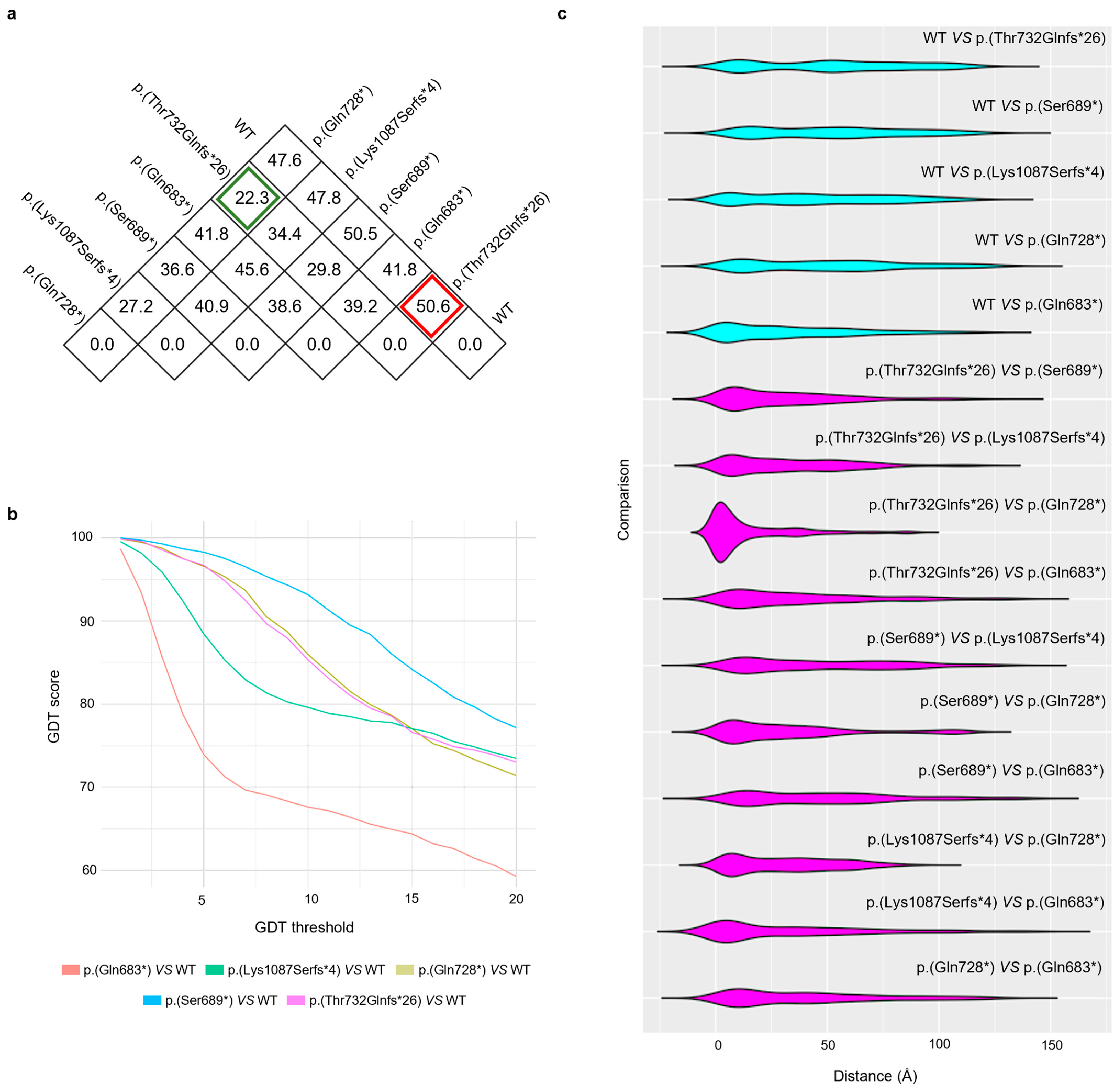
3.3. Protein Inference
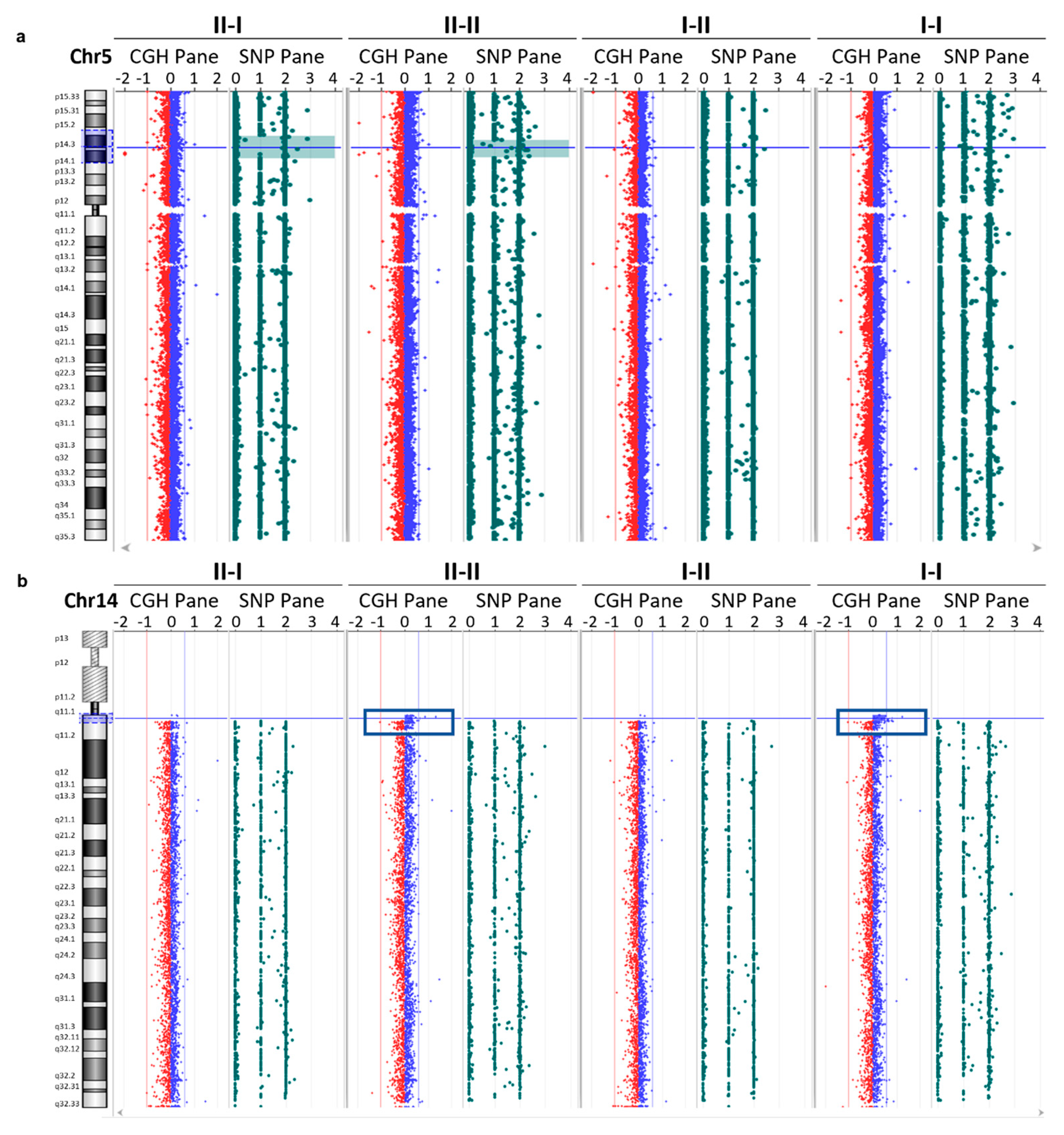
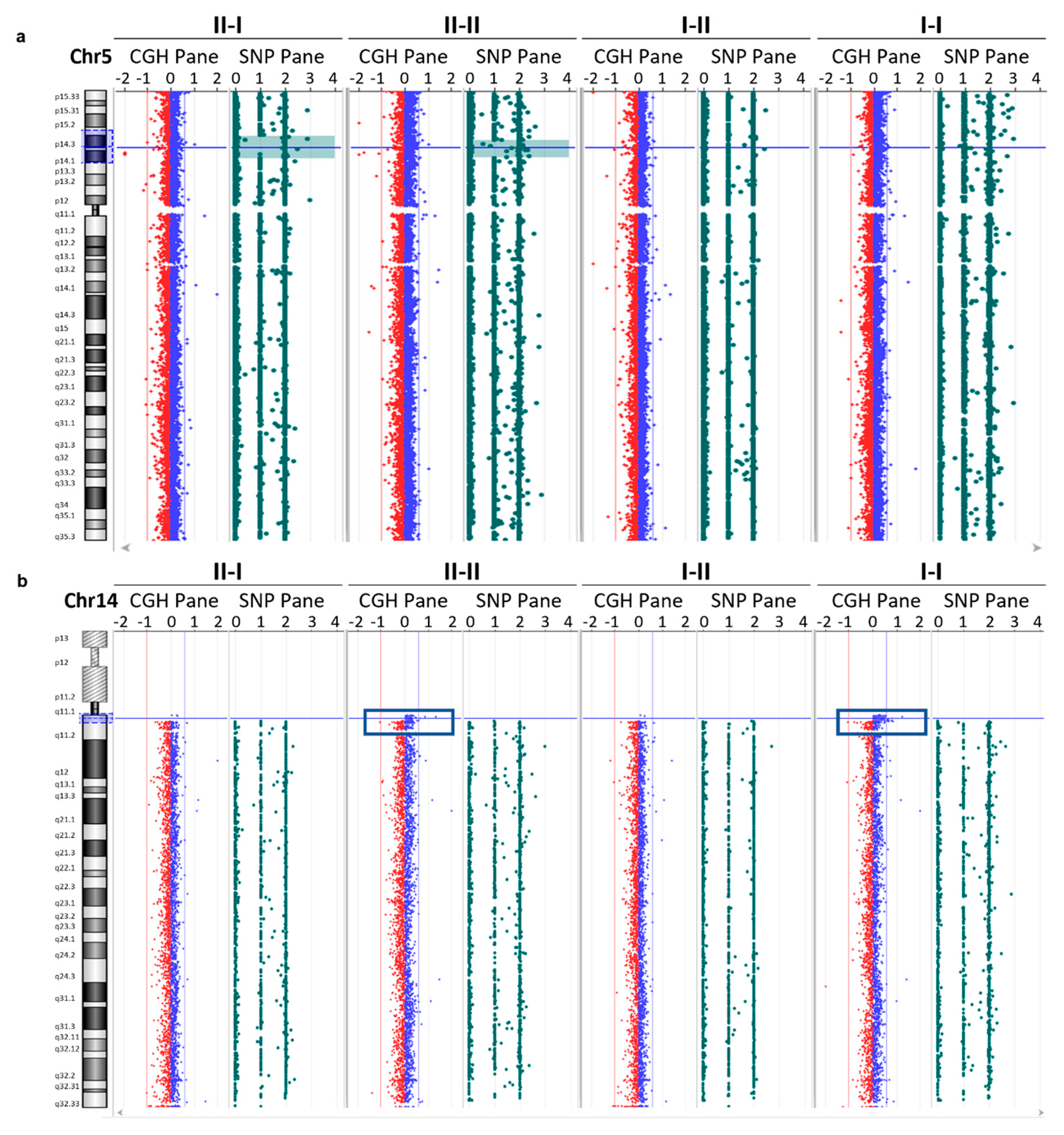
3.4. Array CGH Analysis
3.5. Gene Expression Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
- AlphaFold, https://alphafold.ebi.ac.uk/ (accessed on 26 October 2023)
- PDB, https://www.rcsb.org/ (accessed on 26 October 2023)
- PDB70, https://wwwuser.gwdg.de/~compbiol/data/hhsuite/databases/hhsuite_dbs/ (accessed on 26 October 2023)
- BFD, https://bfd.mmseqs.com/ (accessed on 26 October 2023)
- MGnify, https://www.ebi.ac.uk/metagenomics (accessed on 26 October 2023)
- Primer3Plus, https://primer3.org/ (accessed on 16 August 2022)
- UniRef30 (FKA UniClust30), https://uniclust.mmseqs.com/ (accessed on 26 October 2023)
- UniRef90, https://www.uniprot.org/help/uniref (accessed on 26 October 2023)
- BioPython, http://www.biopython.org (accessed on 26 October 2023)
- GnomAD, https://gnomad.broadinstitute.org/ (accessed on 5 April 2024)
- Clinvar, https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 5 April 2024)
- LOVD, https://www.lovd.nl/ (accessed on 5 April 2024)
References
- Martínez-Frías, M.L.; Arroyo Carrera, I.; Muñoz-Delgado, N.J.; Nieto Conde, C.; Rodríguez-Pinilla, E.; Urioste Azcorra, M.; Omeñaca Teres, F.; García Alix, A. The Adams-Oliver Syndrome in Spain: The Epidemiological Aspects. An. Esp. Pediatr. 1996, 45, 57–61. [Google Scholar] [PubMed]
- Morales, O.L.; Díaz, J.M.; Montoya, J.H. Adams-Oliver Syndrome and Associated Complications: Report of a Family in Colombia and Review of the Literature. Biomedica 2022, 42, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Meester, J.A.N.; Sukalo, M.; Schröder, K.C.; Schanze, D.; Baynam, G.; Borck, G.; Bramswig, N.C.; Duman, D.; Gilbert-Dussardier, B.; Holder-Espinasse, M.; et al. Elucidating the Genetic Architecture of Adams-Oliver Syndrome in a Large European Cohort. Hum. Mutat. 2018, 39, 1246–1261. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.J. Notch Signalling: A Simple Pathway Becomes Complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Sakaidani, Y.; Ichiyanagi, N.; Saito, C.; Nomura, T.; Ito, M.; Nishio, Y.; Nadano, D.; Matsuda, T.; Furukawa, K.; Okajima, T. O-Linked-N-Acetylglucosamine Modification of Mammalian Notch Receptors by an Atypical O-GlcNAc Transferase Eogt1. Biochem. Biophys. Res. Commun. 2012, 419, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Southgate, L.; Machado, R.D.; Snape, K.M.; Primeau, M.; Dafou, D.; Ruddy, D.M.; Branney, P.A.; Fisher, M.; Lee, G.J.; Simpson, M.A.; et al. Gain-of-Function Mutations of ARHGAP31, a Cdc42/Rac1 GTPase Regulator, Cause Syndromic Cutis Aplasia and Limb Anomalies. Am. J. Hum. Genet. 2011, 88, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Isrie, M.; Wuyts, W.; Van Esch, H.; Devriendt, K. Isolated Terminal Limb Reduction Defects: Extending the Clinical Spectrum of Adams-Oliver Syndrome and ARHGAP31 Mutations. Am. J. Med. Genet. A 2014, 164A, 1576–1579. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Durkie, M.; Cassidy, E.; Berry, I.; Owens, M.; Turnbull, C.; Scott, R.H.; Taylor, R.W.; Deans, Z.C.; Ellard, S.; Baple, E.L.; et al. ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2024. In Royal Devon University Healthcare NHS Foundation Trust, Exeter, EX2 5DW. 5; Division of Genetics and Epidemiology: London, UK, 2024; Volume 12. [Google Scholar]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical Standards for the Interpretation and Reporting of Constitutional Copy-Number Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate Normalization of Real-Time Quantitative RT-PCR Data by Geometric Averaging of Multiple Internal Control Genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Bisong, E. Google Colaboratory. In Building Machine Learning and Deep Learning Models on Google Cloud Platform; Apress: Berkeley, CA, USA, 2019; pp. 59–64. [Google Scholar]
- Lefter, M.; Vis, J.K.; Vermaat, M.; den Dunnen, J.T.; Taschner, P.E.M.; Laros, J.F.J. Mutalyzer 2: Next Generation HGVS Nomenclature Checker. Bioinformatics 2021, 37, 2811–2817. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Abagyan, R. Methods of Protein Structure Comparison. In Homology Modeling; Series of Methods in Molecular Biology; Springer Science + Business Media, LLC: Humana Totowa, NJ, USA, 2012; Volume 857, pp. 231–257. [Google Scholar]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern Web App for 3D Visualization and Analysis of Large Biomolecular Structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2004, 32, 115D–D119. [Google Scholar] [CrossRef]
- Tian, H.; Chu, F.; Li, Y.; Xu, M.; Li, W.; Li, C. Synergistic effects of rare variants of ARHGAP31 and FBLN1 in vitro in terminal transverse limb defects. Front. Genet. 2022, 13, 946854. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Caron, C.; DeGeer, J.; Fournier, P.; Duquette, P.M.; Luangrath, V.; Ishii, H.; Karimzadeh, F.; Lamarche-Vane, N.; Royal, I. CdGAP/ARHGAP31, a Cdc42/Rac1 GTPase regulator, is critical for vascular development and VEGF-mediated angiogenesis. Sci. Rep. 2016, 6, 27485. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, D.; Yamada, A.; Amano, T.; Yasuhara, R.; Kimura, A.; Sakahara, M.; Tsumaki, N.; Takeda, S.; Tamura, M.; Nakamura, M.; et al. Essential Mesenchymal Role of Small GTPase Rac1 in Interdigital Programmed Cell Death during Limb Development. Dev. Biol. 2009, 335, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, R.; Yamada, A.; Suzuki, D.; Iimura, T.; Kassai, H.; Harada, T.; Tsukasaki, M.; Yamamoto, G.; Tachikawa, T.; Nakao, K.; et al. Cdc42 Is Required for Chondrogenesis and Interdigital Programmed Cell Death during Limb Development. Mech. Dev. 2012, 129, 38–50. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| II-I | I-I | II-II | |
|---|---|---|---|
| Personal data | |||
| Date of birth | 3 June 1989 | 31 August 1955 | 30 July 1985 |
| Parenthood | Proband | Father | Brother |
| Objective examination | |||
| Gender | F | M | M |
| Height (cm) | 156 | 168 | 170 |
| Foot abnormalities | +++ | + | ++ |
| Hand abnormalities | + | - | - |
| Cranial alterations | + | - | - |
| Instrumental examination | |||
| Foot’s radiography | AD | NA | NA |
| Electrocardiogram | AND | AND | AND |
| Sweat chloride test | AND | NA | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santaniello, C.; Faversani, A.; Corsaro, L.; Melloni, G.; Motta, S.; Mandorino, E.; Sacco, D.; Stioui, S.; Ferrara, F.; Barteselli, D.; et al. Characterization of a New Variant in ARHGAP31 Probably Involved in Adams–Oliver Syndrome in a Family with a Variable Phenotypic Spectrum. Genes 2024, 15, 536. https://doi.org/10.3390/genes15050536
Santaniello C, Faversani A, Corsaro L, Melloni G, Motta S, Mandorino E, Sacco D, Stioui S, Ferrara F, Barteselli D, et al. Characterization of a New Variant in ARHGAP31 Probably Involved in Adams–Oliver Syndrome in a Family with a Variable Phenotypic Spectrum. Genes. 2024; 15(5):536. https://doi.org/10.3390/genes15050536
Chicago/Turabian StyleSantaniello, Carlo, Alice Faversani, Luigi Corsaro, Giulia Melloni, Silvia Motta, Elena Mandorino, Davide Sacco, Sabine Stioui, Fulvio Ferrara, Davide Barteselli, and et al. 2024. "Characterization of a New Variant in ARHGAP31 Probably Involved in Adams–Oliver Syndrome in a Family with a Variable Phenotypic Spectrum" Genes 15, no. 5: 536. https://doi.org/10.3390/genes15050536