
Statistical Genetic Approaches to Investigate Genotype-by-Environment Interaction: Review and Novel Extension of Models
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Statistical Genetic Models and Inference
2.1.1. Polygenic Model
2.1.2. Modeling the Genotype-by-Environment Interaction for Discrete and Continuous Environments
2.1.3. Joint Genotype-by-Environment Interaction for Discrete and Continuous Environments
2.2. Statistical Inferential Theory
2.3. Comparison of Sex-Specific Additive Genetic Variance Functions
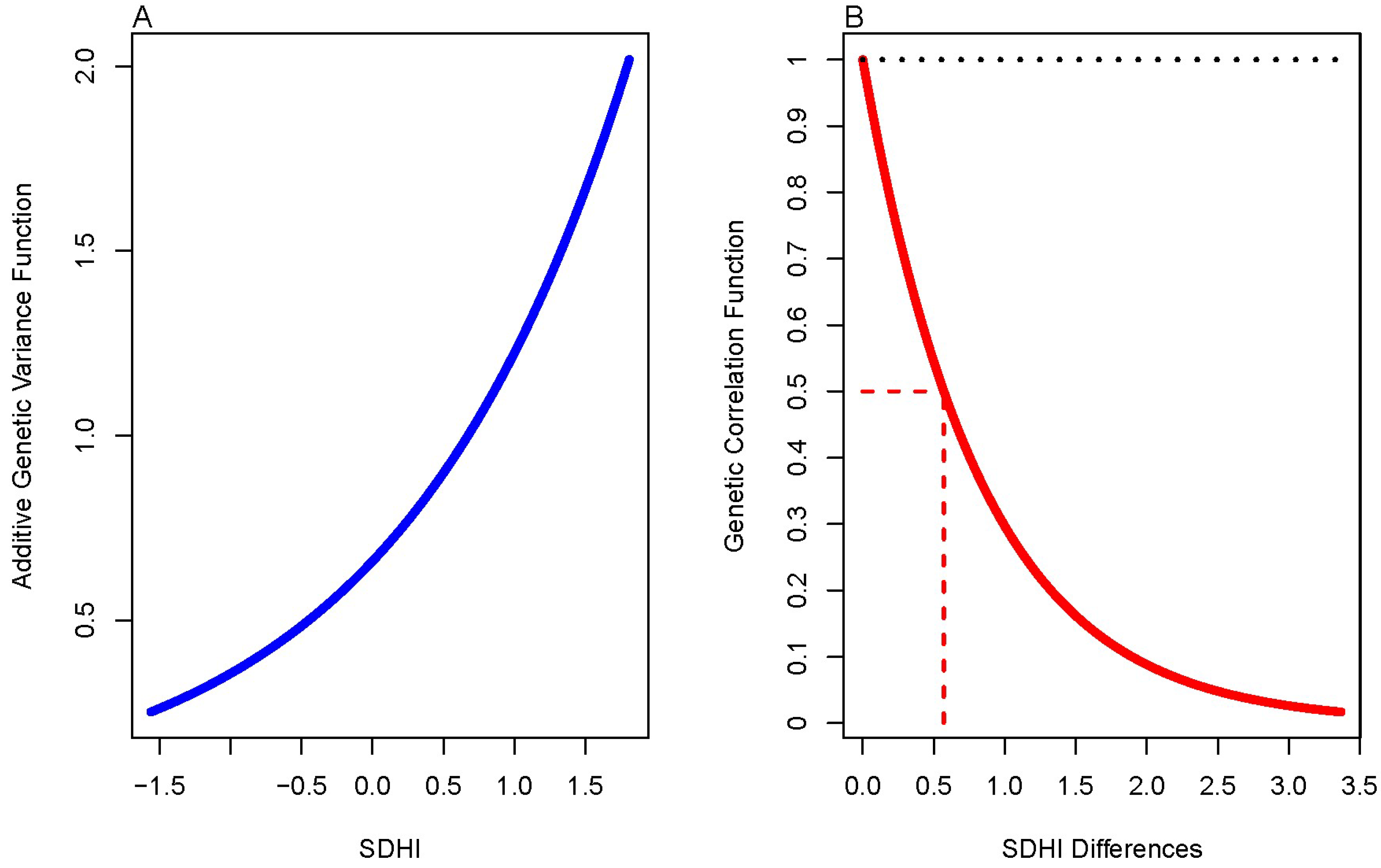
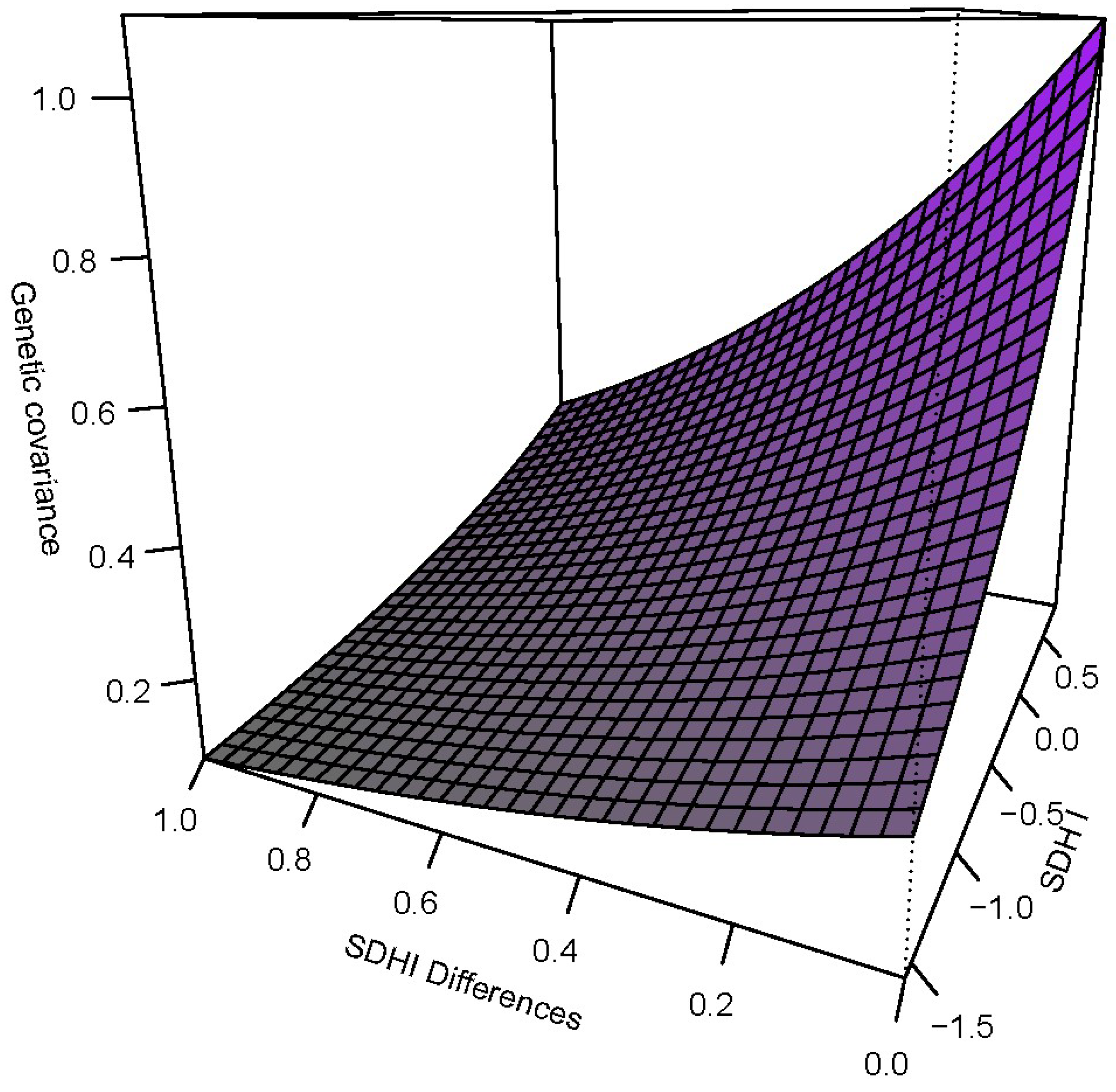
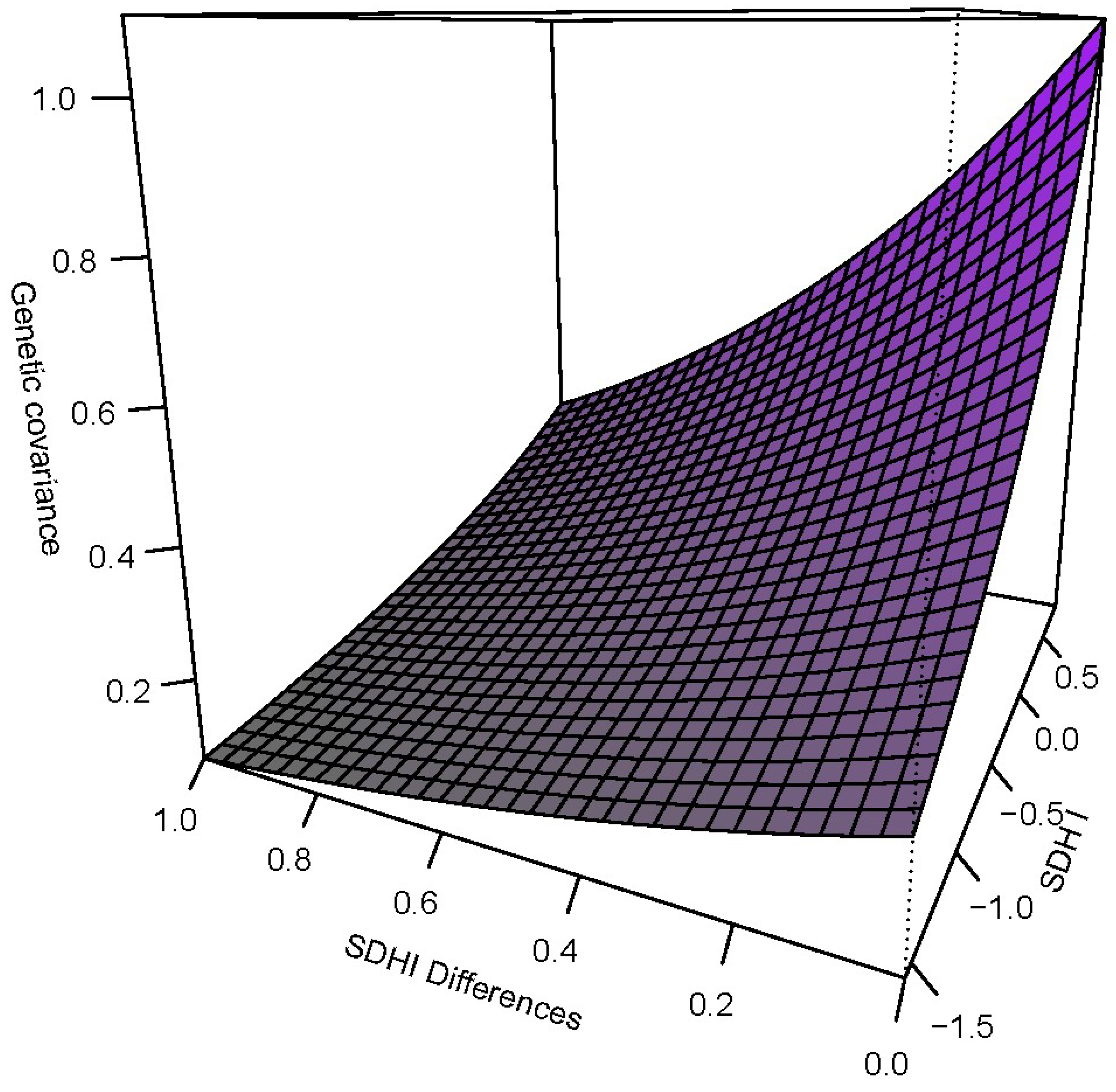
3. Results
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A. On the Statistical Genetic Models
Appendix A.1. Polygenic Model
Appendix A.2. Modeling the Genotype-by-Environment Interaction for Discrete and Continuous Environments
Appendix A.3. Joint Genotype-by-Environment Interaction for Discrete and Continuous Environments
References
- Blangero, J. Statistical genetic approaches to human adaptability. Hum. Biol. 2009, 81, 523–546. [Google Scholar] [CrossRef] [PubMed]
- Viel, K.R.; Warren, D.M.; Buil, A.; Dyer, T.D.; Howard, T.E.; Almasy, L. A comparison of discrete versus continuous environment in a variance components-based linkage analysis of the COGA data. BMC Genet. 2005, 6, S57. [Google Scholar] [CrossRef] [PubMed]
- Avery, C.L.; Freedman, B.I.; Kraja, A.T.; Borecki, I.B.; Miller, M.B.; Pankow, J.S.; Arnett, D.; Lewis, C.E.; Myers, R.H.; Hunt, S.C.; et al. Genotype-by-sex interaction in the aetiology of type 2 diabetes mellitus: Support for sex-specific quantitative trait loci in Hypertension Genetic Epidemiology Network participants. Diabetologia 2006, 49, 2329–2336. [Google Scholar] [CrossRef] [PubMed]
- Winnier, D.A.; Rainwater, D.L.; Cole, S.A.; Williams, J.T.; Dyer, T.D.; Blangero, J.; MacCluer, J.W.; Mahaney, M.C. Sex-specific QTL effects on variation in paraoxonase 1 (PON1) activity in Mexican Americans. Genet. Epidemiol. 2007, 31, 66–74. [Google Scholar] [CrossRef] [PubMed]
- North, K.E.; Franceschini, N.; Borecki, I.B.; Gu, C.C.; Heiss, G.; Province, M.A.; Arnett, D.K.; Lewis, C.E.; Miller, M.B.; Myers, R.H.; et al. Genotype-by-Sex Interaction on Fasting Insulin Concentration: The HyperGEN Study. Diabetes 2007, 56, 137–142. [Google Scholar] [CrossRef]
- Diego, V.P.; de Chaves, R.N.; Blangero, J.; de Souza, M.C.; Santos, D.; Gomes, T.N.; dos Santos, F.K.; Garganta, R.; Katzmarzyk, P.T.; Maia, J.A. Sex-specific genetic effects in physical activity: Results from a quantitative genetic analysis. BMC Med. Genet. 2015, 16, 58. [Google Scholar] [CrossRef]
- Poveda, A.; Chen, Y.; Brändström, A.; Engberg, E.; Hallmans, G.; Johansson, I.; Renström, F.; Kurbasic, A.; Franks, P.W. The heritable basis of gene-environment interactions in cardiometabolic traits. Diabetologia 2017, 60, 442–452. [Google Scholar] [CrossRef]
- Diego, V.P.; Manusov, E.G.; Mao, X.; Almeida, M.; Peralta, J.M.; Curran, J.E.; Mahaney, M.C.; Göring, H.; Blangero, J.; Williams-Blangero, S. Metabolic syndrome traits exhibit genotype-by-environment interaction in relation to socioeconomic status in the Mexican American family heart study. Front. Genet. 2024, 15, 1240462. [Google Scholar] [CrossRef]
- Glahn, D.C.; Kent, J.W., Jr.; Sprooten, E.; Diego, V.P.; Winkler, A.M.; Curran, J.E.; McKay, D.R.; Knowles, E.E.; Carless, M.A.; Göring, H.H.; et al. Genetic basis of neurocognitive decline and reduced white-matter integrity in normal human brain aging. Proc. Natl. Acad. Sci. USA 2013, 110, 19006–19011. [Google Scholar] [CrossRef]
- Arya, R.; Farook, V.S.; Fowler, S.P.; Puppala, S.; Chittoor, G.; Resendez, R.G.; Mummidi, S.; Vanamala, J.; Almasy, L.; Curran, J.E.; et al. Genetic and environmental (physical fitness and sedentary activity) interaction effects on cardiometabolic risk factors in Mexican American children and adolescents. Genet. Epidemiol. 2018, 42, 378–393. [Google Scholar] [CrossRef]
- Pittner, K.; Bakermans-Kranenburg, M.J.; Alink, L.R.A.; Buisman, R.S.M.; van den Berg, L.J.M.; Block, L.H.C.G.C.C.-d.; Voorthuis, A.; Elzinga, B.M.; Lindenberg, J.; Tollenaar, M.S.; et al. Estimating the Heritability of Experiencing Child Maltreatment in an Extended Family Design. Child Maltreatment 2020, 25, 289–299. [Google Scholar] [CrossRef]
- Manusov, E.G.; Diego, V.P.; Sheikh, K.; Laston, S.; Blangero, J.; Williams-Blangero, S. Non-alcoholic Fatty Liver Disease and Depression: Evidence for Genotype × Environment Interaction in Mexican Americans. Front. Psychiatry 2022, 13, 936052. [Google Scholar] [CrossRef] [PubMed]
- Diego, V.P.; Manusov, E.G.; Mao, X.; Curran, J.E.; Göring, H.; Almeida, M.; Mahaney, M.C.; Peralta, J.M.; Blangero, J.; Williams-Blangero, S. Genotype-by-socioeconomic status interaction influences heart disease risk scores and carotid artery thickness in Mexican Americans: The predominant role of education in comparison to household income and socioeconomic index. Front. Genet. 2023, 14, 1132110. [Google Scholar] [CrossRef]
- Marmot, M.G.; Rose, G.; Shipley, M.; Hamilton, P.J. Employment grade and coronary heart disease in British civil servants. J. Epidemiol. Community Health 1978, 32, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Marmot, M.G.; Stansfeld, S.; Patel, C.; North, F.; Head, J.; White, I.; Brunner, E.; Feeney, A.; Marmot, M.G.; Smith, G.D. Health inequalities among British civil servants: The Whitehall II study. Lancet 1991, 337, 1387–1393. [Google Scholar] [CrossRef]
- Langenberg, C.; Shipley, M.J.; Batty, G.D.; Marmot, M.G. Adult Socioeconomic Position and the Association Between Height and Coronary Heart Disease Mortality: Findings From 33 Years of Follow-Up in the Whitehall Study. Am. J. Public Health 2005, 95, 628–632. [Google Scholar] [CrossRef]
- Stringhini, S.; Batty, G.D.; Bovet, P.; Shipley, M.J.; Marmot, M.G.; Kumari, M.; Tabak, A.G.; Kivimäki, M. Association of Lifecourse Socioeconomic Status with Chronic Inflammation and Type 2 Diabetes Risk: The Whitehall II Prospective Cohort Study. PLoS Med. 2013, 10, e1001479. [Google Scholar] [CrossRef] [PubMed]
- Shim, R.S.; Ye, J.; Baltrus, P.; Fry-Johnson, Y.; Daniels, E.; Rust, G. Racial/ethnic disparities, social support, and depression: Examining a social determinant of mental health. Ethn. Dis. 2012, 22, 15–20. [Google Scholar]
- Allen, J.; Balfour, R.; Bell, R.; Marmot, M. Social determinants of mental health. Int. Rev. Psychiatry 2014, 26, 392–407. [Google Scholar] [CrossRef]
- Shim, R.; Koplan, C.; Langheim, F.J.; Manseau, M.W.; Powers, R.A.; Compton, M.T. The social determinants of mental health: An overview and call to action. Psychiatr. Ann. 2014, 44, 22–26. [Google Scholar] [CrossRef]
- World Health Organization. Social Determinants of Mental Health; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Carod-Artal, F.J. Social Determinants of Mental Health. In Global Mental Health: Prevention and Promotion; Bährer-Kohler, S., Carod-Artal, F.J., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 33–46. [Google Scholar] [CrossRef]
- Alegría, M.; NeMoyer, A.; Falgàs Bagué, I.; Wang, Y.; Alvarez, K. Social Determinants of Mental Health: Where We Are and Where We Need to Go. Curr. Psychiatry Rep. 2018, 20, 95. [Google Scholar] [CrossRef]
- Jeste, D.V.; Koh, S.; Pender, V.B. Perspective: Social Determinants of Mental Health for the New Decade of Healthy Aging. Am. J. Geriatr. Psychiatry 2022, 30, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Jeste, D.V.; Pender, V.B. Social Determinants of Mental Health: Recommendations for Research, Training, Practice, and Policy. JAMA Psychiatry 2022, 79, 283–284. [Google Scholar] [CrossRef] [PubMed]
- Shim, R.S.; Compton, M.T. The Social Determinants of Mental Health: Psychiatrists’ Roles in Addressing Discrimination and Food Insecurity. Focus 2020, 18, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Tafet, G.E.; Nemeroff, C.B. The links between stress and depression: Psychoneuroendocrinological, genetic, and environmental interactions. J. Neuropsychiatry Clin. Neurosci. 2016, 28, 77–88. [Google Scholar] [CrossRef] [PubMed]
- LeMoult, J. From stress to depression: Bringing together cognitive and biological science. Curr. Dir. Psychol. Sci. 2020, 29, 592–598. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S.; Akil, H. Revisiting the stress concept: Implications for affective disorders. J. Neurosci. 2020, 40, 12–21. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S. Protective and damaging effects of stress mediators: Central role of the brain. Dialogues Clin. Neurosci. 2022, 8, 367–381. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S. Physiology and Neurobiology of Stress and Adaptation: Central Role of the Brain. Physiol. Rev. 2007, 87, 873–904. [Google Scholar] [CrossRef]
- McEwen, B.S. Neurobiological and Systemic Effects of Chronic Stress. Chronic Stress 2017, 1, 2470547017692328. [Google Scholar] [CrossRef]
- Wilkinson, R.G.; Marmot, M. Social Determinants of Health: The Solid Facts; World Health Organization: Geneva, Switzerland, 2003. [Google Scholar]
- Kang, H.-J.; Park, Y.; Yoo, K.-H.; Kim, K.-T.; Kim, E.-S.; Kim, J.-W.; Kim, S.-W.; Shin, I.-S.; Yoon, J.-S.; Kim, J.H. Sex differences in the genetic architecture of depression. Sci. Rep. 2020, 10, 9927. [Google Scholar] [CrossRef] [PubMed]
- Labonté, B.; Engmann, O.; Purushothaman, I.; Menard, C.; Wang, J.; Tan, C.; Scarpa, J.R.; Moy, G.; Loh, Y.-H.E.; Cahill, M. Sex-specific transcriptional signatures in human depression. Nat. Med. 2017, 23, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Seney, M.L.; Glausier, J.; Sibille, E. Large-scale transcriptomics studies provide insight into sex differences in depression. Biol. Psychiatry 2022, 91, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Silveira, P.P.; Pokhvisneva, I.; Howard, D.M.; Meaney, M.J. A sex-specific genome-wide association study of depression phenotypes in UK Biobank. Mol. Psychiatry 2023, 28, 2469–2479. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kim, D.; Cho, Y. Exploratory Factor Analysis of the Beck Anxiety Inventory and the Beck Depression Inventory-II in a Psychiatric Outpatient Population. J. Korean Med. Sci. 2018, 33, e128. [Google Scholar] [CrossRef]
- Penley, J.A.; Wiebe, J.S.; Nwosu, A. Psychometric properties of the Spanish Beck Depression Inventory-II in a medical sample. Psychol. Assess. 2003, 15, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Wiebe, J.S.; Penley, J.A. A psychometric comparison of the Beck Depression Inventory-II in English and Spanish. Psychol. Assess. 2005, 17, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.P.; Gorenstein, C. Psychometric properties of the Beck Depression Inventory-II: A comprehensive review. Braz. J. Psychiatry 2013, 35, 416–431. [Google Scholar] [CrossRef] [PubMed]
- Billioux, A.; Verlander, K.; Anthony, S.; Alley, D. Standardized screening for health-related social needs in clinical settings: The accountable health communities screening tool. In NAM Perspectives; National Academy of Medicine: Washington, DC, USA, 2017. [Google Scholar]
- Lange, K. Mathematical and Statistical Methods for Genetic Analysis; Springer: Berlin/Heidelberg, Germany, 2002; Volume 488. [Google Scholar]
- Blangero, J.; Diego, V.P.; Dyer, T.D.; Almeida, M.; Peralta, J.; Kent, J.W., Jr.; Williams, J.T.; Almasy, L.; Göring, H.H. A kernel of truth: Statistical advances in polygenic variance component models for complex human pedigrees. Adv. Genet. 2013, 81, 1–31. [Google Scholar] [CrossRef]
- Diego, V.P.; Kent, J.W.; Blangero, J. Familial Studies: Genetic Inferences. In International Encyclopedia of the Social & Behavioral Sciences, 2nd ed.; Wright, J.D., Ed.; Elsevier: Oxford, UK, 2015; pp. 715–724. [Google Scholar] [CrossRef]
- Sorensen, D.; Gianola, D. Likelihood, Bayesian and MCMC Methods in Quantitative Genetics; Springer: Berlin/Heidelberg, Germany, 2002. [Google Scholar]
- Quillen, E.E.; Voruganti, V.S.; Chittoor, G.; Rubicz, R.; Peralta, J.M.; Almeida, M.A.; Kent, J.W., Jr.; Diego, V.P.; Dyer, T.D.; Comuzzie, A.G.; et al. Evaluation of estimated genetic values and their application to genome-wide investigation of systolic blood pressure. BMC Proc. 2014, 8, S66. [Google Scholar] [CrossRef]
- Almasy, L.; Blangero, J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am. J. Hum. Genet. 1998, 62, 1198–1211. [Google Scholar] [CrossRef] [PubMed]
- Self, S.G.; Liang, K.-Y. Asymptotic Properties of Maximum Likelihood Estimators and Likelihood Ratio Tests under Nonstandard Conditions. J. Am. Stat. Assoc. 1987, 82, 605–610. [Google Scholar] [CrossRef]
- Dominicus, A.; Skrondal, A.; Gjessing, H.K.; Pedersen, N.L.; Palmgren, J. Likelihood ratio tests in behavioral genetics: Problems and solutions. Behav. Genet. 2006, 36, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M. A note on the asymptotic distribution of likelihood ratio tests to test variance components. Twin. Res. Hum. Genet. 2006, 9, 490–495. [Google Scholar] [CrossRef] [PubMed]
- DasGupta, A. Asymptotic Theory of Statistics and Probability; Springer: Berlin/Heidelberg, Germany, 2008; Volume 180. [Google Scholar]
- Giampaoli, V.; Singer, J.M. Likelihood ratio tests for variance components in linear mixed models. J. Stat. Plan. Inference 2009, 139, 1435–1448. [Google Scholar] [CrossRef]
- Azzalini, A. Statistical Inference Based on the Likelihood; Routledge: London, UK, 2017. [Google Scholar]
- Edwards, A.W.F. Likelihood: An Account of the Statistical Concept of Likelihood and Its Application to Scientific Inference; Johns Hopkins University Press: Baltimore, MA, USA, 1992. [Google Scholar]
- Held, L.; Bové, D.S. Likelihood and Bayesian Inference. Statistics for Biology and Health; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Pawitan, Y. All Likelihood: Statistical Modelling and Inference Using Likelihood; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Severini, T.A. Likelihood Methods in Statistics; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Neudecker, H.; Trenkler, G. On the approximate variance of a nonlinear function of random variables. In Contributions to Probability and Statistics: Applications and Challenges; World Scientific: Singapore, 2006; pp. 172–177. [Google Scholar]
- Nakagawa, S.; Cuthill, I.C. Effect size, confidence interval and statistical significance: A practical guide for biologists. Biol. Rev. 2007, 82, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Julious, S.A. Using confidence intervals around individual means to assess statistical significance between two means. Pharm. Stat. 2004, 3, 217–222. [Google Scholar] [CrossRef]
- Knol, M.J.; Pestman, W.R.; Grobbee, D.E. The (mis)use of overlap of confidence intervals to assess effect modification. Eur. J. Epidemiol. 2011, 26, 253–254. [Google Scholar] [CrossRef] [PubMed]
- Maghsoodloo, S.; Huang, C.-Y. Comparing the overlapping of two independent confidence intervals with a single confidence interval for two normal population parameters. J. Stat. Plan. Inference 2010, 140, 3295–3305. [Google Scholar] [CrossRef]
- Molenberghs, G.; Verbeke, G. Likelihood Ratio, Score, and Wald Tests in a Constrained Parameter Space. Am. Stat. 2007, 61, 22–27. [Google Scholar] [CrossRef]
- Elston, R.; Rao, D. Statistical modeling and analysis in human genetics. Annu. Rev. Biophys. Bioeng. 1978, 7, 253–286. [Google Scholar] [CrossRef] [PubMed]
- Lange, K.; Westlake, J.; Spence, M.A. Extensions to pedigree analysis III. Variance components by the scoring method. Ann. Hum. Genet. 1976, 39, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, M.; Heckman, N. A quantitative genetic model for growth, shape, reaction norms, and other infinite-dimensional characters. J. Math. Biol. 1989, 27, 429–450. [Google Scholar] [CrossRef] [PubMed]
- Pletcher, S.D.; Geyer, C.J. The genetic analysis of age-dependent traits: Modeling the character process. Genetics 1999, 153, 825–835. [Google Scholar] [CrossRef]
- Jaffrézic, F.; Pletcher, S.D. Statistical models for estimating the genetic basis of repeated measures and other function-valued traits. Genetics 2000, 156, 913–922. [Google Scholar] [CrossRef]
- Pletcher, S.D.; Jaffrézic, F. Generalized character process models: Estimating the genetic basis of traits that cannot be observed and that change with age or environmental conditions. Biometrics 2002, 58, 157–162. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Trait | Females N = 389 | Males N = 133 | p-Value | ||
|---|---|---|---|---|---|
| Mean | SD | Mean | SD | ||
| Age | 44.33 | 14.76 | 45.96 | 15.70 | 0.2936 |
| BDI-II | 19.88 | 15.99 | 25.67 | 19.99 | 3.1 × 10−4 |
| AHA HRSN | 0.10 | 0.14 | 0.07 | 0.12 | 0.0025 |
| Trait | Heritability | Standard Error | Sample Size | p-Value |
|---|---|---|---|---|
| BDI-II | 0.37 | 0.14 | 521 | 7.8 × 10−6 |
| AHC HRSN | 0.40 | 0.13 | 521 | 6.6 × 10−4 |
| Trait | Model | Ln Likelihood | Chi-Square | p-Value |
|---|---|---|---|---|
| BDI-II | Polygenic | −247.716 | 36.97529 | 2.8 × 10−8 |
| G × Sex | −229.228 |
| Trait | Model | Ln Likelihood | Chi-Square | p-Value |
|---|---|---|---|---|
| BDI-II | Additive genetic variance homogeneity | −229.317 | 0.178 | 0.67 |
| Residual environmental variance homogeneity | −233.465 | 8.473 | 1.0 × 10−3 | |
| Constrained genetic correlation across sex | −234.850 | 11.240 | 4.0 × 10−4 | |
| G × Sex interaction model | −229.230 |
| Trait | Model | Ln Likelihood | Chi-Square | p-Value |
|---|---|---|---|---|
| BDI-II | Polygenic | −241.401 | 22.026 | 9.6 × 10−6 |
| Red. G × E | −230.388 |
| Trait | Model | Ln Likelihood | Chi-Square | p-Value |
|---|---|---|---|---|
| BDI-II | Constrained genetic slope | −237.646 | 14.517 | 1.3 × 10−4 |
| Constrained environmental slope | --- | --- | --- | |
| Constrained genetic correlation decay | −233.509 | 6.245 | 6.2 × 10−3 | |
| Red. G × E interaction model | −230.388 |
| Trait | Model | Ln Likelihood | Chi-Square | p-Value |
|---|---|---|---|---|
| BDI-II | G × Sex | −229.228 | 42.73996 | 1.8 × 10−8 |
| Red. G × E | −207.858 |
| Trait | Model | Ln Likelihood | Chi-Square | p-Value |
|---|---|---|---|---|
| BDI-II | Constrained genetic slope in females | −208.182 | 0.647 | 0.42 |
| Constrained environmental slope in females | −207.942 | 0.169 | 0.68 | |
| Constrained genetic correlation decay in females | −208.576 | 1.436 | 0.12 | |
| Constrained genetic slope in males | −216.248 | 16.780 | 4.2 × 10−5 | |
| Constrained environmental slope in males | --- | --- | --- | |
| Constrained genetic correlation decay in males | −208.28 | 0.84433 | 0.18 | |
| Constrained across-sex genetic correlation | −211.642 | 7.568402 | 2.0 × 10−3 | |
| Red. G × E interaction model | −207.858 |
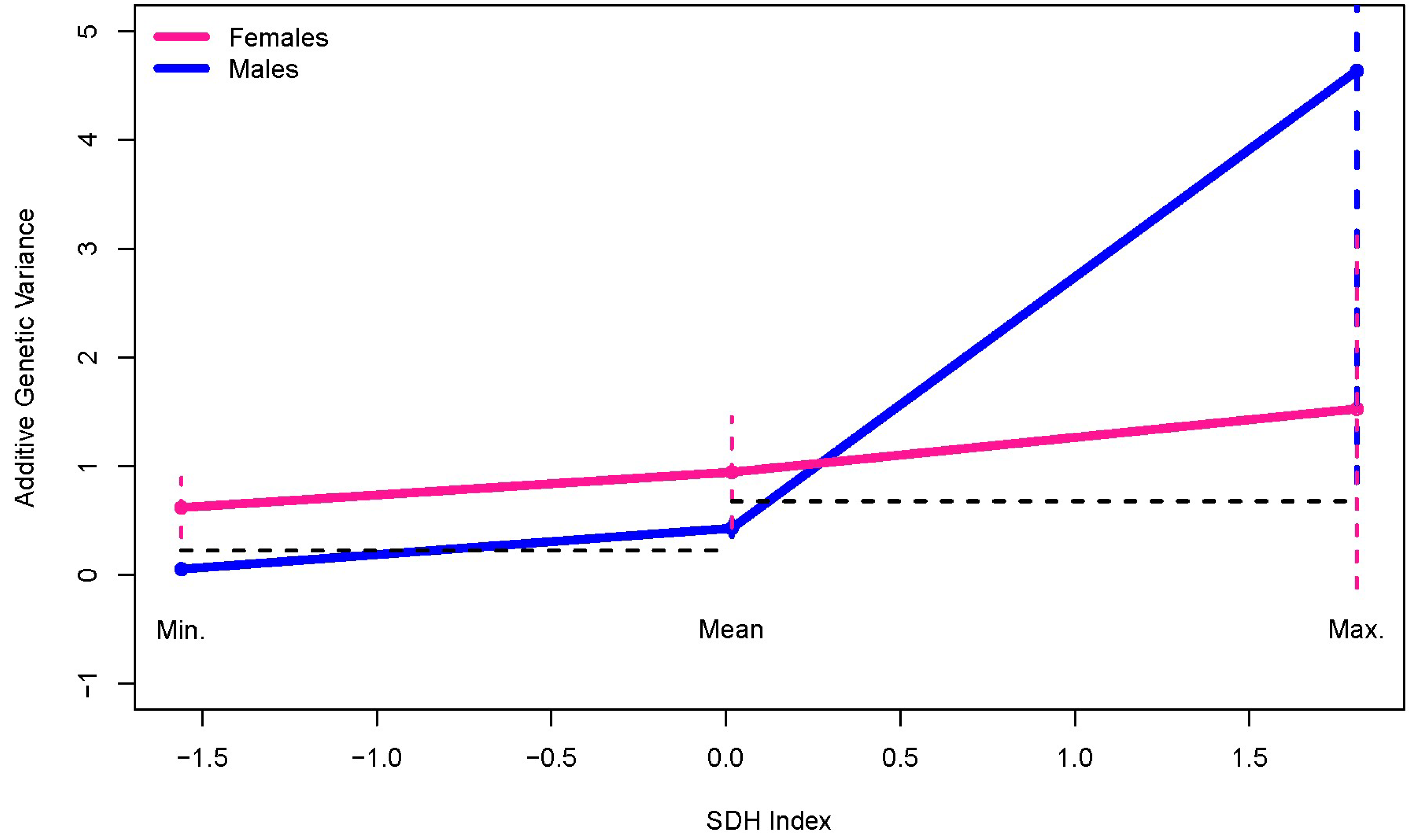
| Sex | Additive Genetic Variance | Adjusted Lower Bound * | Adjusted Upper Bound | Wald Statistic | p-Value |
|---|---|---|---|---|---|
| Min. SDHI | |||||
| Females | 0.618 | 0.347 | 0.890 | 6.527 | 0.011 |
| Males | 0.053 | 0.013 | 0.092 | ||
| Mean SDHI | |||||
| Females | 0.944 | 0.437 | 1.451 | 1.550 | 0.213 |
| Males | 0.429 | 0.354 | 0.503 | ||
| Max. SDHI | |||||
| Females | 1.5253 | −0.1185 | 3.169 | 0.6485 | 0.4206 |
| Males | 4.6378 | 0.8509 | 8.4247 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diego, V.P.; Manusov, E.G.; Almeida, M.; Laston, S.; Ortiz, D.; Blangero, J.; Williams-Blangero, S. Statistical Genetic Approaches to Investigate Genotype-by-Environment Interaction: Review and Novel Extension of Models. Genes 2024, 15, 547. https://doi.org/10.3390/genes15050547
Diego VP, Manusov EG, Almeida M, Laston S, Ortiz D, Blangero J, Williams-Blangero S. Statistical Genetic Approaches to Investigate Genotype-by-Environment Interaction: Review and Novel Extension of Models. Genes. 2024; 15(5):547. https://doi.org/10.3390/genes15050547
Chicago/Turabian StyleDiego, Vincent P., Eron G. Manusov, Marcio Almeida, Sandra Laston, David Ortiz, John Blangero, and Sarah Williams-Blangero. 2024. "Statistical Genetic Approaches to Investigate Genotype-by-Environment Interaction: Review and Novel Extension of Models" Genes 15, no. 5: 547. https://doi.org/10.3390/genes15050547