Heritability of Radiation Response in Lung Cancer Families

Abstract
:1. Introduction
2. Results and Discussion

{kind=link}
{kind=link}
| Generations | Composition of family | Total | |||||
|---|---|---|---|---|---|---|---|
| Parents | LC case | Siblings | Partners | Offspring | |||
| 1 (unsuitable for the estimation of heritability) | -- | 1 | -- | -- | -- | 1 | 9 |
| -- | 1 | -- | ≥1 | -- | 1 | ||
| -- | 1 | ≥1 | ≥1 | -- | 7 | ||
| 2 | -- | -- | ≥1 | ≥1 | ≥1 | 1 | 133 |
| -- | 1 | -- | -- | ≥1 | 4 | ||
| -- | 1 | -- | ≥1 | ≥1 | 9 | ||
| -- | 1 | ≥1 | ≥1 | ≥1 | 31 | ||
| 1 | -- | -- | ≥1 | -- | 1 | ||
| 1 | 1 | -- | -- | -- | 6 | ||
| 1 | 1 | -- | ≥1 | -- | 35 | ||
| 1 | 1 | ≥1 | ≥1 | -- | 3 | ||
| 2 | 1 | -- | -- | -- | 18 | ||
| 2 | 1 | -- | ≥1 | -- | 24 | ||
| 2 | 1 | ≥1 | -- | -- | 1 | ||
| 3 | 1 | 1 | -- | -- | ≥1 | 1 | 35 |
| 1 | 1 | -- | ≥1 | ≥1 | 10 | ||
| 1 | 1 | ≥1 | -- | ≥1 | 1 | ||
| 1 | 1 | ≥1 | ≥1 | ≥1 | 9 | ||
| 2 | 1 | -- | -- | ≥1 | 1 | ||
| 2 | 1 | -- | ≥1 | ≥1 | 3 | ||
| 2 | 1 | ≥1 | -- | ≥1 | 3 | ||
| 2 | 1 | ≥1 | ≥1 | ≥1 | 7 | ||
| Total | 177 | ||||||
2.1. DNA Damage and DNA Repair

2.2. Impact of Age, Sex and Smoking

2.3. Heritability
 ) [34,34,35,36]. Depending on the availability of parental outcome measures we performed mid-parent-offspring regression or one parent-offspring regression. Because the COMET assay is an experiment affected by numerous noisy or confounding factors [26], we assessed the effect of the experimental design onto the estimation of heritability. Hereby we introduced a falsity-correction for
) [34,34,35,36]. Depending on the availability of parental outcome measures we performed mid-parent-offspring regression or one parent-offspring regression. Because the COMET assay is an experiment affected by numerous noisy or confounding factors [26], we assessed the effect of the experimental design onto the estimation of heritability. Hereby we introduced a falsity-correction for  considering pseudo-families generated from independent controls of a control cohort (KORA). All controls within a single trial were randomly grouped in pseudo-families, where the status as pseudo-parent or pseudo-offspring was assigned by chance. Heritability was estimated as described above in each of 3000 repetitions. The mean estimate was considered as falsity
considering pseudo-families generated from independent controls of a control cohort (KORA). All controls within a single trial were randomly grouped in pseudo-families, where the status as pseudo-parent or pseudo-offspring was assigned by chance. Heritability was estimated as described above in each of 3000 repetitions. The mean estimate was considered as falsity 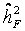 induced by study and assay design. For internal validation we investigated spouse-correlation in terms of spearman’s rank correlation of lnOTM between partners and parents, siblings and the index person respectively, separately for basal damage, induced damage and residual damage.
induced by study and assay design. For internal validation we investigated spouse-correlation in terms of spearman’s rank correlation of lnOTM between partners and parents, siblings and the index person respectively, separately for basal damage, induced damage and residual damage.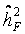 = 20%, which was used as falsity-correction factor. Hence, an adjustment of the primary outcome parameter by repeated measurement of a single human blood donor (an identical human reference sample used in every experimental set) is insufficient in clearing interfering design or assay specific factors [26]. However, heritability in pseudo-families ranges from 4% to 28% (Table 2), indicating the methodological difficulties for an appropriate correction. This range might be due to experimental variability at short time points of DNA repair and the large proportion of white noise in the radiation induced damage [26].
= 20%, which was used as falsity-correction factor. Hence, an adjustment of the primary outcome parameter by repeated measurement of a single human blood donor (an identical human reference sample used in every experimental set) is insufficient in clearing interfering design or assay specific factors [26]. However, heritability in pseudo-families ranges from 4% to 28% (Table 2), indicating the methodological difficulties for an appropriate correction. This range might be due to experimental variability at short time points of DNA repair and the large proportion of white noise in the radiation induced damage [26].| Falsity-correction
| Nominal heritability
 | Corrected heritability
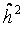 | |
|---|---|---|---|
| pseudo-families | LC-families | LC-families | |
| Basal damage | 23% | 77% (63%–91% ) | 70% (52%–88%) |
| Initial damage | 28% | 75%(62%–88% ) | 65% (47%–83%) |
| Repair 10 min | 22% | 56% (45%–67% ) | 44% (29%–58%) |
| Repair 30 min | 18% | 34% (24%–43% ) | 20% (07%–30%) |
| Repair 60 min | 4% | 50% (36%–65% ) | 48% (33%–64%) |
3. Experimental Section
3.1. Study Groups
3.2. Isolation of Lymphocytes, Irradiation and DNA Repair
3.3. Exposure Level and Batch Processing, COMET Assay, Image Acquisition and COMET Analysis
3.4. Measures for DNA Damage
3.5. Impact of Age, Sex and Smoking
3.6. Estimation of Heritability
[34,36,42]. Depending on the availability of parental outcome measures we performed mid-parent-offspring regression (  if measures of both parents were available) or one parent-offspring regression (
if measures of both parents were available) or one parent-offspring regression ( 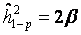 if measures of only one parents were available), where
if measures of only one parents were available), where  is the regression coefficient of a linear regression, adjusted for the offspring’s mean age and mean gender and the value of a human control to adjust for inter-assay variability. Both heritability estimates were than pooled by
is the regression coefficient of a linear regression, adjusted for the offspring’s mean age and mean gender and the value of a human control to adjust for inter-assay variability. Both heritability estimates were than pooled by  (weighted mean) with weights equal to the inverse of the estimated sample variance
(weighted mean) with weights equal to the inverse of the estimated sample variance  and
and 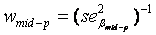 . The standard error of was calculated as
. The standard error of was calculated as 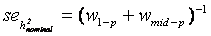 [43].considering independent controls. All controls within a single trial were randomly grouped in pseudo-families, where the status as pseudo-parent or pseudo-offspring was assigned by chance. Heritability was estimated as described above in each of 3000 repetitions. The mean estimate was considered as falsity induced by study and assay design. Depending on the range of values for heritability spans from
[43].considering independent controls. All controls within a single trial were randomly grouped in pseudo-families, where the status as pseudo-parent or pseudo-offspring was assigned by chance. Heritability was estimated as described above in each of 3000 repetitions. The mean estimate was considered as falsity induced by study and assay design. Depending on the range of values for heritability spans from 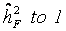 (instead of
(instead of  ) the final estimate is given by
) the final estimate is given by  .
.4. Conclusions
Acknowledgments
Conflict of interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar]
- Ezzati, M.; Henley, S.J.; Lopez, A.D.; Thun, M.J. Role of smoking in global and regional cancer epidemiology: current patterns and data needs. Int. J. Cancer 2005, 116, 963–971. [Google Scholar]
- Samet, J.M.; Avila-Tang, E.; Boffetta, P.; Hannan, L.M.; Olivo-Marston, S.; Michael J. Thun, M.J.; Rudin, C.M. Lung cancer in never smokers: clinical epidemiology and environmental risk factors. Clin. Cancer Res. 2009, 15, 5626–5645. [Google Scholar]
- Wakelee, H.A.; Chang, E.T.; Gomez, S.L.; Keegan, T.H.; Feskanich, D.; Clarke, C.A.; Holmberg, L.; Yong, L.C.; Kolonel, L.N.; Gould, M.K.; West, D.W. Lung cancer incidence in never smokers. J. Clin. Oncol. 2007, 25, 472–478. [Google Scholar]
- Landi, M.T.; Chatterjee, N.; Yu, K.; Goldin, L.R.; Goldstein, A.M.; Rotunno, M.; Mirabello, L.; Jacobs, K.; Wheeler, W.; Yeager, M. A genome-wide association study of lung cancer identifies a region of chromosome 5p15 associated with risk for adenocarcinoma. Am. J. Hum. Genet. 2009, 85, 679–691. [Google Scholar]
- Truong, T.; Hung, R.J.; Amos, C.I.; Wu, X.; Bickeböller, H.; Rosenberger, A.; Sauter, W.; Illig, T.; Wichmann, H.-E.; Risch, A. Replication of lung cancer susceptibility loci at chromosomes 15q25, 5p15, and 6p21: A pooled analysis from the International Lung Cancer Consortium. J. Natl. Cancer Inst. 2010, 102, 959–971. [Google Scholar] [CrossRef]
- Hung, R.J.; Christiani, D.C.; Risch, A.; Popanda, O.; Haugen, A.; Zienolddiny, S.; Benhamou, S.; Bouchardy, C.; Lan, Q.; Spitz, M.R. International Lung Cancer Consortium: Pooled analysis of sequence variants in DNA repair and cell cycle pathways. Cancer Epidemiol. Biomark. Prev. 2008, 17, 3081–3089. [Google Scholar]
- Kreuzer, M.; Kreienbrock, L.; Gerken, M.; Heinrich, J.; Bruske-Hohlfeld, I.; Muller, K.-M.; Wichmann, H.E. Risk factors for lung cancer in young adults. Am. J. Epidemiol. 1998, 147, 1028–1037. [Google Scholar]
- Schwartz, A.G.; Yang, P.; Swanson, G.M. Familial risk of lung cancer among nonsmokers and their relatives. Am. J. Epidemiol. 1996, 144, 554–562. [Google Scholar]
- Sellers, T.A.; Chen, P.L.; Potter, J.D.; Bailey-Wilson, J.E.; Rothschild, H.; Elston, R.C. Segregation analysis of smoking-associated malignancies: Evidence for Mendelian inheritance. Am. J. Med. Genet. 1994, 52, 308–314. [Google Scholar]
- Bailey-Wilson, J.E.; Sellers, T.A.; Elston, R.C.; Evens, C.C.; Rothschild, H. Evidence for a major gene effect in early-onset lung cancer. J. La State Med. Soc. 1993, 145, 157–162. [Google Scholar]
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Iliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukkala, E.; Skytthe, A.; Hemminki, K. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000, 343, 78–85. [Google Scholar] [CrossRef]
- Braun, M.M.; Caporaso, N.E.; Page, W.F.; Hoover, R.N. Genetic component of lung cancer: Cohort study of twins. Lancet 1994, 344, 440–443. [Google Scholar]
- Scott, D. Chromosomal radiosensitivity and low penetrance predisposition to cancer. Cytogenet. Genome Res. 2004, 104, 365–370. [Google Scholar]
- Berwick, M.; Vineis, P. Markers of DNA repair and susceptibility to cancer in humans: An epidemiologic review. J. Natl. Cancer Inst. 2000, 92, 874–897. [Google Scholar]
- Lavin, M.F.; Kozlov, S. ATM activation and DNA damage response. Cell Cycle 2007, 6, 931–942. [Google Scholar]
- Kobayashi, J.; Antoccia, A.; Tauchi, H.; Matsuura, S.; Komatsu, K. NBS1 and its functional role in the DNA damage response. DNA Repair (Amst) 2004, 3, 855–861. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Voronova, N.V.; Chistiakov, P.A. Genetic variations in DNA repair genes, radiosensitivity to cancer and susceptibility to acute tissue reactions in radiotherapy-treated cancer patients. Acta Oncol. 2008, 47, 809–824. [Google Scholar]
- O'Driscoll, M.; Gennery, A.R.; Seidel, J.; Concannon, P.; Jeggo, P.A. An overview of three new disorders associated with genetic instability: LIG4 syndrome, RS-SCID and ATR-Seckel syndrome. DNA Repair (Amst) 2004, 3, 1227–1235. [Google Scholar] [CrossRef]
- Wu, X.; Spitz, M.R.; Amos, C.I.; Lin, J.; Shao, L.; Gu, J.; de Andrade, M.; Neal, L.; Benowitz, N.L.; Peter, G.; Shields, P.G.; Swan, G.E. Mutagen sensitivity has high heritability: Evidence from a twin study. Cancer Res. 2006, 66, 5993–5996. [Google Scholar]
- Roberts, S.A.; Spreadborough, A.R.; Bulman, B.; Barber, J.B.; Evans, D.G.; Scott, D. Heritability of cellular radiosensitivity: A marker of low-penetrance predisposition genes in breast cancer? Am. J. Hum. Genet. 1999, 65, 784–794. [Google Scholar] [CrossRef]
- Borgmann, K.; Haeberle, D.; Doerk, T.; Busjahn, A.; Stephan, G.; Dikomey, E. Genetic determination of chromosomal radiosensitivities in G0- and G2-phase human lymphocytes. Radiother. Oncol. 2007, 83, 196–202. [Google Scholar]
- Surowy, H.; Rinckleb, A.; Luedeke, M.; Stuber, M.; Wecker, A.; Varga, D.; Maier, C.; Hoegel, J.; Vogel, W. Heritability of baseline and induced micronucleus frequencies. Mutagenesis 2011, 26, 111–117. [Google Scholar]
- Schmitz, A.; Bayer, J.; Dechamps, N.; Goldin, L.; Thomas, G. Heritability of susceptibility to ionizing radiation-induced apoptosis of human lymphocyte subpopulations. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 1169–1177. [Google Scholar]
- Rajeswari, N.; Ahuja, Y.R.; Malini, U.; Chandrashekar, S.; Balakrishna, N.; Rao, K.V.; Khar, A. Risk assessment in first degree female relatives of breast cancer patients using the alkaline Comet assay. Carcinogenesis 2000, 21, 557–561. [Google Scholar]
- Rosenberger, A.; Rossler, U.; Hornhardt, S.; Sauter, W.; Bickeböller, H.; Wichmann, H.-E.; Gomolka, M. Validation of a fully automated COMET assay: 1.75 million single cells measured over a 5 year period. DNA Repair (Amst) 2011, 10, 322–337. [Google Scholar] [CrossRef]
- Collins, A.R.; Oscoz, A.A.; Brunborg, G.; Gaivão, I.; Giovannelli, L.; Kruszewski, M.; Smith, C.C.; Stetina, R. The comet assay: Topical issues. Mutagenesis 2008, 23, 143–151. [Google Scholar]
- Moller, P. The alkaline comet assay: Towards validation in biomonitoring of DNA damaging exposures. Basic Clin. Pharmacol. Toxicol. 2006, 98, 336–345. [Google Scholar]
- Statistisches Bundesamt Deustschland. Raucherquote Erwachsene 1995,1999,2003,2005,2009. 3-8-2011. GENESIS-Tabelle.
- Hornhardt, S.; Rößler, U.; Sauter, W.; Rosenberger, A.; Illig, T.; Bickeböller, H.; Wichmann, H.E.; Gomolka, M. Genetic factors of individual radiation sensitivity in lung cancer families. 2011; in preparation. [Google Scholar]
- Gomolka, M.; Rossler, U.; Hornhardt, S.; Walsh, L.; Panzer, W.; Schmid, E. Measurement of the initial levels of DNA damage in human lymphocytes induced by 29 kV X rays (mammography X rays) relative to 220 kV X rays and gamma rays. Radiat. Res. 2005, 163, 510–519. [Google Scholar]
- Tice, R.R.; Agurell, E.; Anderson, D.; Burlinson, B.; Hartmann, A.; Kobayashi, H.; Miyamae, Y.; Rojas, E.; Ryu, J.C.; Sasaki, Y.F. Single cell gel/comet assay: Guidelines for in vitro and in vivo genetic toxicology testing. Environ. Mol. Mutagen. 2000, 35, 206–221. [Google Scholar]
- Lee, E.; Oh, E.; Lee, J.; Sul, D.; Lee, J. Use of the tail moment of the lymphocytes to evaluate DNA damage in human biomonitoring studies. Toxicol. Sci. 2004, 81, 121–132. [Google Scholar]
- Falconer, D.S.; Mackay, T.F.C. Introduction to Quantitative Genetics, 4th ed; Longman Scientific & Technical: Harlow, Essex, UK, 1996. [Google Scholar]
- Fernandez, G.; Miller, J. Estimation of heritability by parent-offspring regression. TAG Theor. Appl. Genet. 1985, 70, 650–654. [Google Scholar]
- Khoury, M.J.; Beaty, T.H.; Cohen, B.H. Fundamentals of Genetic Epidemiology; Oxford University Press: New York, NY, USA, 1993. [Google Scholar]
- Mittelstrass, K.; Sauter, W.; Rosenberger, A.; Illig, T.; Timofeeva, M.; Klopp, N.; Dienemann, H.; Meese, E.; Sybrecht, G.; Woelke, G. Early onset lung cancer, cigarette smoking and the SNP309 of the murine double minute-2 (MDM2) gene. BMC Cancer 2008, 8, 113. [Google Scholar]
- Sauter, W.; Rosenberger, A.; Beckmann, L.; Kropp, S.; Mittelstrass, K.; Timofeeva, M.; Wölke, G.; Steinwachs, A.; Scheiner, D.; Meese, E. Matrix metalloproteinase 1 (MMP1) is associated with early-onset lung cancer. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1127–1135. [Google Scholar]
- Holle, R.; Happich, M.; Lowel, H.; Wichmann, H.E. KORA-a research platform for population based health research. Gesundheitswesen 2005, 67, S19–S25. [Google Scholar]
- Wichmann, H.E.; Gieger, C.; Illig, T. KORA-gen--resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen 2005, 67, S26–S30. [Google Scholar]
- Rosenberger, A.; Illig, T.; Korb, K.; Klopp, N.; Zietemann, V.; Wölke, G.; Meese, E.; Sybrecht, G.; Kronenberg, F.; Cebulla, M. Do genetic factors protect for early onset lung cancer? A case control study before the age of 50 years. BMC Cancer 2008, 8, 60. [Google Scholar]
- Fernandez, G.; Miller, J. Estimation of heritability by parent-offspring regression. TAG Theor. Appl. Genet. 1985, 70, 650–654. [Google Scholar]
- Petitti, D.B. Meta-Analysis, Decision Analysis, and Cost-Effectiveness Analysis Methods for Quantitative Synthesis in Medicine, 2nd ed; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rosenberger, A.; Rössler, U.; Hornhardt, S.; Sauter, W.; Bickeböller, H.; Wichmann, H.-E.; Gomolka, M. Heritability of Radiation Response in Lung Cancer Families. Genes 2012, 3, 248-260. https://doi.org/10.3390/genes3020248
Rosenberger A, Rössler U, Hornhardt S, Sauter W, Bickeböller H, Wichmann H-E, Gomolka M. Heritability of Radiation Response in Lung Cancer Families. Genes. 2012; 3(2):248-260. https://doi.org/10.3390/genes3020248
Chicago/Turabian StyleRosenberger, Albert, Ute Rössler, Sabine Hornhardt, Wiebke Sauter, Heike Bickeböller, H.-Erich Wichmann, and Maria Gomolka. 2012. "Heritability of Radiation Response in Lung Cancer Families" Genes 3, no. 2: 248-260. https://doi.org/10.3390/genes3020248
APA StyleRosenberger, A., Rössler, U., Hornhardt, S., Sauter, W., Bickeböller, H., Wichmann, H.-E., & Gomolka, M. (2012). Heritability of Radiation Response in Lung Cancer Families. Genes, 3(2), 248-260. https://doi.org/10.3390/genes3020248