Genome-Wide Analysis of Alpharetroviral Integration in Human Hematopoietic Stem/Progenitor Cells

Abstract
:1. Introduction
2. Experimental
2.1. Vectors and Cells
3. Results and Discussion
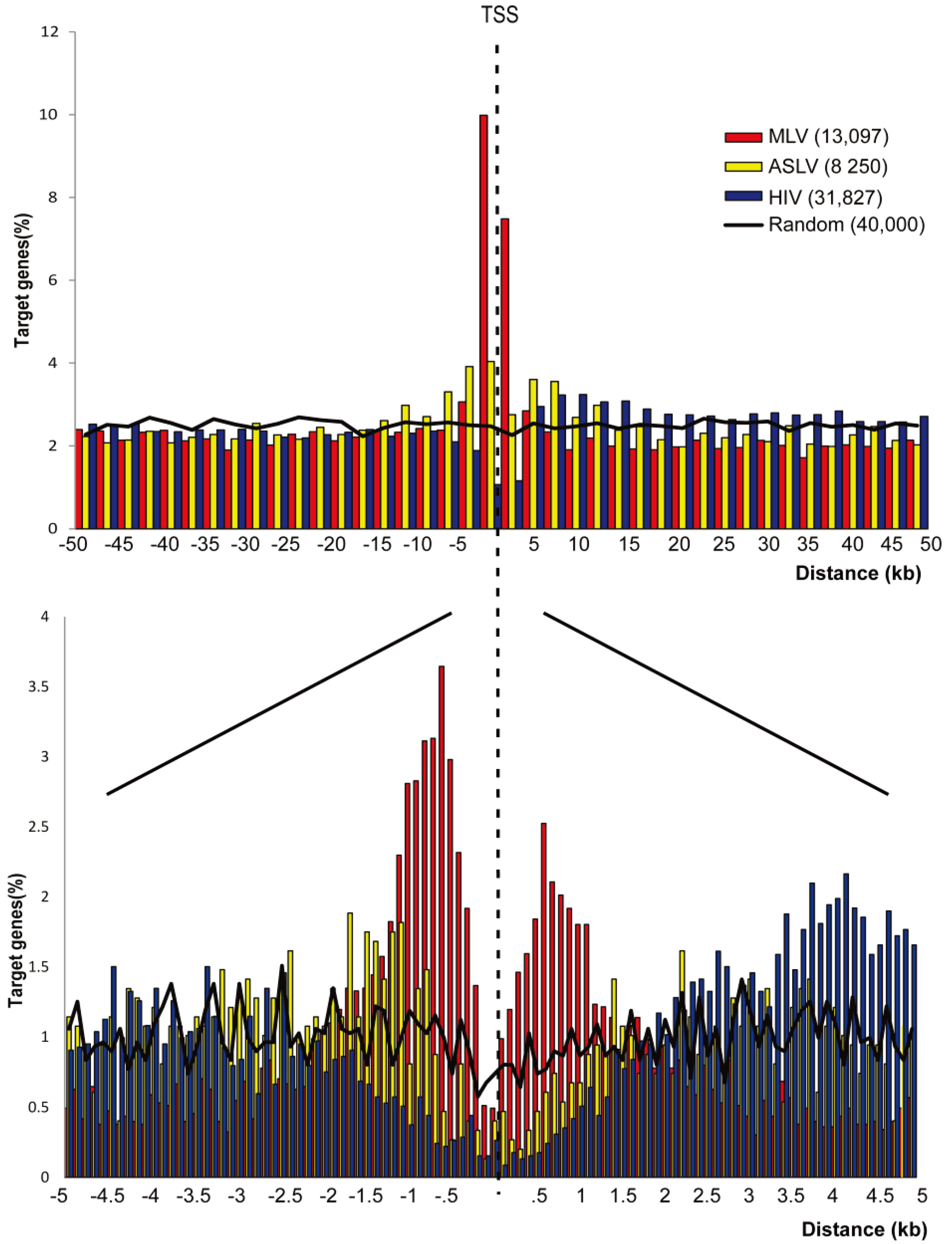
3.1. SIN-ASLV Vectors Exhibit an Almost Random Integration Profile in the Genome of Human CD34+ HSPCs

{kind=link}
{kind=link}
{kind=link}
| Vector | Intergenic (%) | TSS-proximal (%) | Intragenic (%) | CpG islands (%) | CNCs (%) | TFBS (%) | Total integrations |
|---|---|---|---|---|---|---|---|
| SIN-ASLV | 43.55 * | 6.97 | 49.48 | 2.84 | 5.49 | 55.70 | 8250 |
| SIN-MLV | 34.36 * | 23.38 * | 42.26 | 17.68 * | 8.42 | 69.95 * | 13,097 |
| SIN-HIV | 19.78 * | 3.45 | 76.77 * | 1.23 | 4.58 | 54.61 | 31,827 |
| Random | 56.26 | 3.16 | 40.58 | 1.76 | 6.05 | 51.01 | 40,000 |

| Vector | Repetitive elements (%) | LINEs (%) | SINEs (%) | Satellites (%) | LTRs (%) | Others (%) |
|---|---|---|---|---|---|---|
| SIN-ASLV (8,899) | 51.29 | 19.50 | 19.77 | 0.43 | 6.26 | 5.35 |
| SIN-MLV (13,606) | 37.75 | 10.96 | 17.26 | 0.01 | 5.09 | 4.42 |
| SIN-HIV (32,964) | 45.78 | 19.45 | 16.86 | 0.05 | 4.08 | 5.35 |
| Random (40,000) | 50.96 | 21.27 | 14.69 | 0.35 | 9.57 | 5.10 |
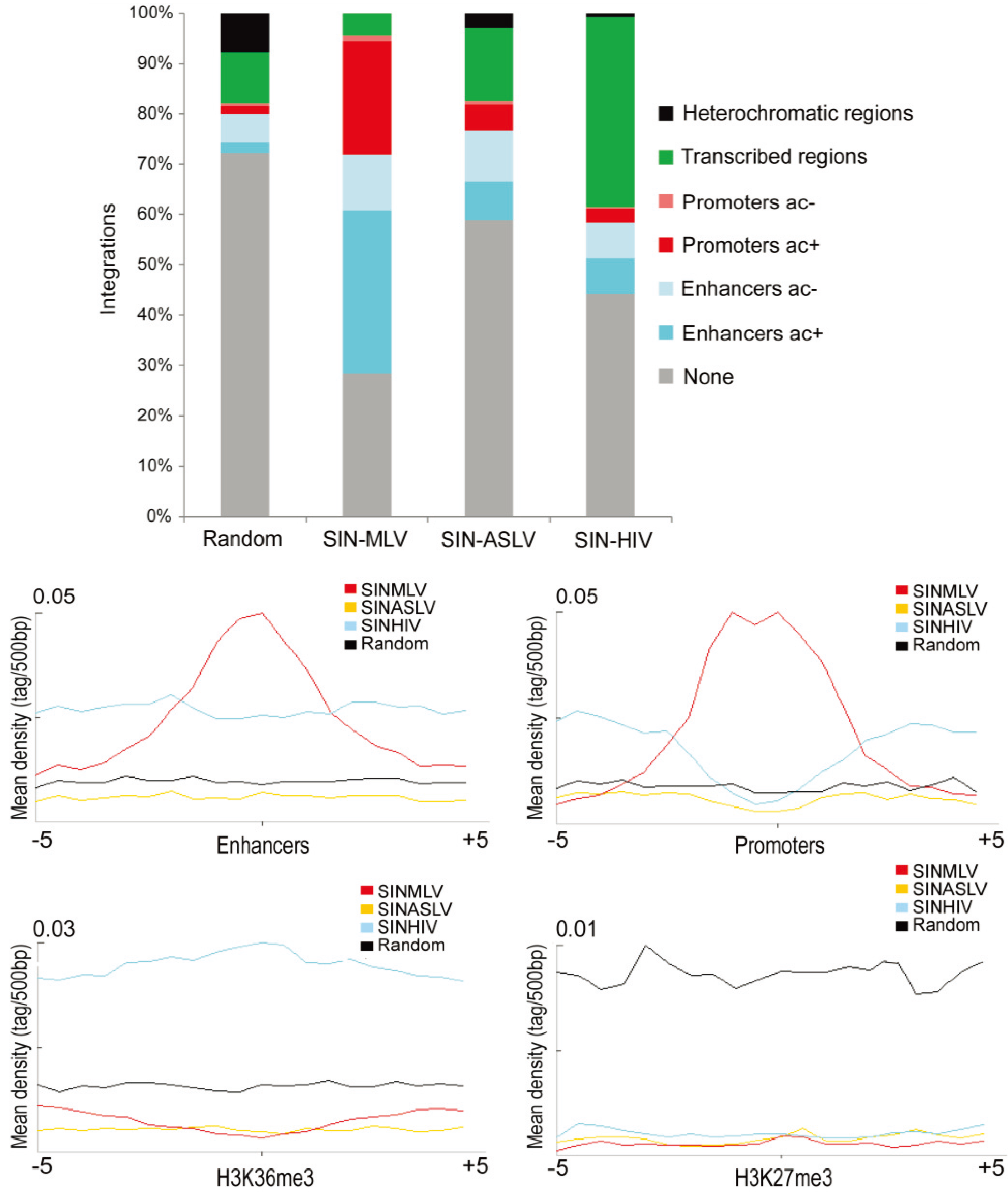
3.2. SIN-ASLV Integration Is Not Associated with Epigenetically-Defined Functional Genomic Regions

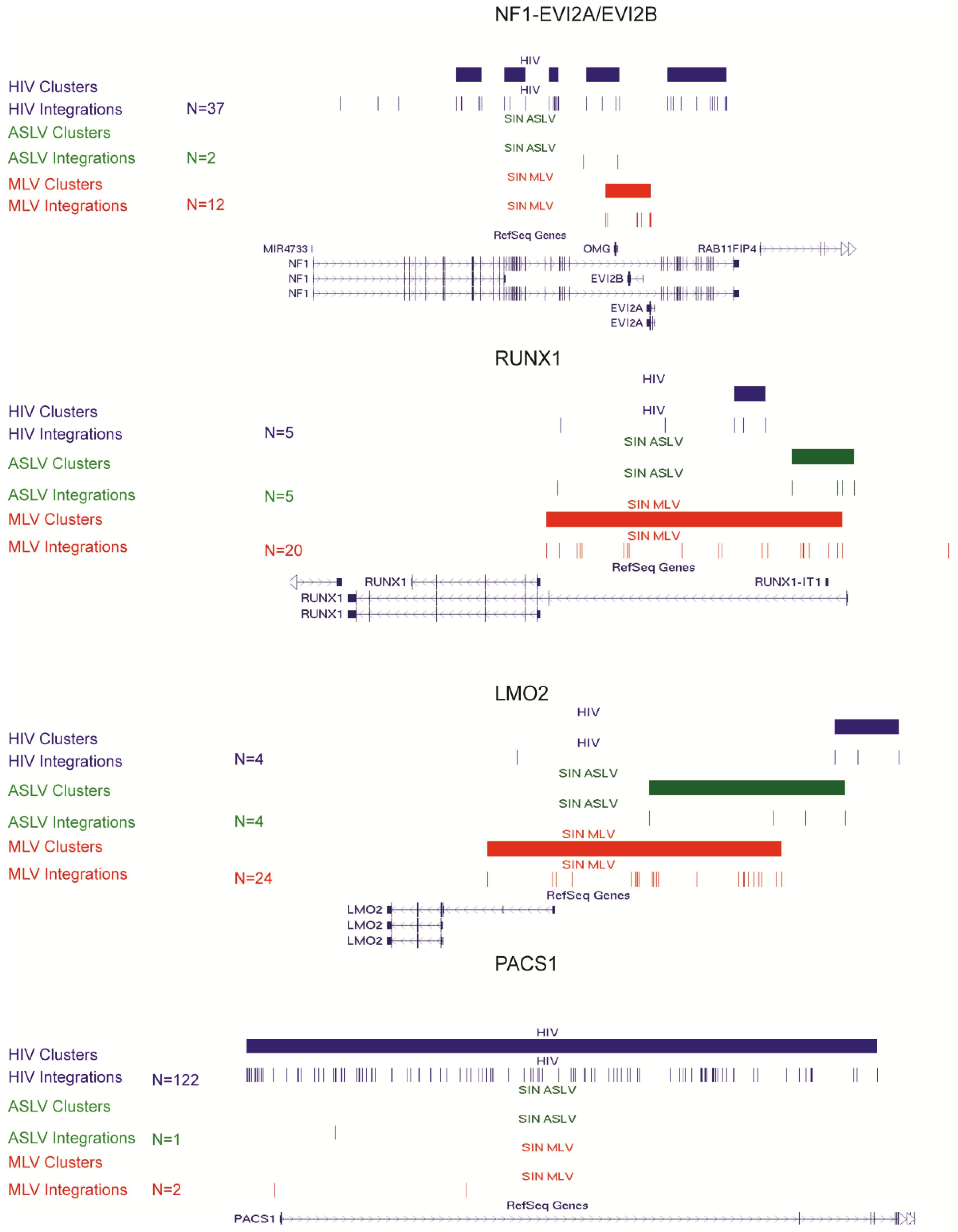
3.3. SIN-ASLV Vector Integrations Are Not Clustered in the Human Genome
| SIN-MLV (13,097) | SIN-ASLV (8250) | SIN-HIV (31,827) | |
|---|---|---|---|
| Clusters | 1415 | 484 | 2724 |
| Integrations in clusters (%) | 56 | 21 | 51 |
| Average cluster dimension | 5.1 | 3.6 | 5.9 |

4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hacein-Bey-Abina, S.; Le Deist, F.; Carlier, F.; Bouneaud, C.; Hue, C.; de Villartay, J.P.; Thrasher, A.J.; Wulffraat, N.; Sorensen, R.; Dupuis-Girod, S.; et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N. Engl. J. Med. 2002, 346, 1185–1193. [Google Scholar] [CrossRef]
- Ott, M.G.; Schmidt, M.; Schwarzwaelder, K.; Stein, S.; Siler, U.; Koehl, U.; Glimm, H.; Kuhlcke, K.; Schilz, A.; Kunkel, H.; et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRMD16 or SETBP1. Nat. Med. 2006, 12, 401–409. [Google Scholar] [CrossRef]
- Mavilio, F.; Pellegrini, G.; Ferrari, S.; di Nunzio, F.; di Iorio, E.; Recchia, A.; Maruggi, G.; Ferrari, G.; Provasi, E.; Bonini, C.; et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat. Med. 2006, 12, 1397–1402. [Google Scholar]
- Aiuti, A.; Cattaneo, F.; Galimberti, S.; Benninghoff, U.; Cassani, B.; Callegaro, L.; Scaramuzza, S.; Andolfi, G.; Mirolo, M.; Brigida, I.; et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 2009, 360, 447–458. [Google Scholar] [CrossRef]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef]
- Boztug, K.; Schmidt, M.; Schwarzer, A.; Banerjee, P.P.; Diez, I.A.; Dewey, R.A.; Bohm, M.; Nowrouzi, A.; Ball, C.R.; Glimm, H.; et al. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N. Engl. J. Med. 2010, 363, 1918–1927. [Google Scholar] [CrossRef]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013. [Google Scholar] [CrossRef]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 2013. [Google Scholar] [CrossRef]
- Cavazza, A.; Moiani, A.; Mavilio, F. Mechanisms of retroviral integration and mutagenesis. Human Gene Ther. 2013, 24, 119–131. [Google Scholar] [CrossRef]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Ponzoni, M.; Bartholomae, C.; Sergi Sergi, L.; Benedicenti, F.; Ambrosi, A.; di Serio, C.; et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 2006, 24, 687–696. [Google Scholar] [CrossRef]
- Modlich, U.; Navarro, S.; Zychlinski, D.; Maetzig, T.; Knoess, S.; Brugman, M.H.; Schambach, A.; Charrier, S.; Galy, A.; Thrasher, A.J.; et al. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol. Ther. 2009, 17, 1919–1928. [Google Scholar] [CrossRef]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of hsc gene therapy. J. Clin. Invest. 2009, 119, 964–975. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M. Transcription start regions in the human genome are favored targets for mlv integration. Science 2003, 300, 1749–1751. [Google Scholar] [CrossRef]
- Cattoglio, C.; Pellin, D.; Rizzi, E.; Maruggi, G.; Corti, G.; Miselli, F.; Sartori, D.; Guffanti, A.; di Serio, C.; Ambrosi, A.; et al. High-definition mapping of retroviral integration sites identifies active regulatory elements in human multipotent hematopoietic progenitors. Blood 2010, 116, 5507–5517. [Google Scholar] [CrossRef]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Wang, G.P.; Ciuffi, A.; Leipzig, J.; Berry, C.C.; Bushman, F.D. HIV integration site selection: Analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 2007, 17, 1186–1194. [Google Scholar] [CrossRef]
- Barr, S.D.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F.D. Integration targeting by avian sarcoma-leukosis virus and human immunodeficiency virus in the chicken genome. J. Virol. 2005, 79, 12035–12044. [Google Scholar] [CrossRef]
- Hu, J.; Renaud, G.; Gomes, T.J.; Ferris, A.; Hendrie, P.C.; Donahue, R.E.; Hughes, S.H.; Wolfsberg, T.G.; Russell, D.W.; Dunbar, C.E. Reduced genotoxicity of avian sarcoma leukosis virus vectors in rhesus long-term repopulating cells compared to standard murine retrovirus vectors. Mol. Ther. 2008, 16, 1617–1623. [Google Scholar] [CrossRef]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004, 2, e234. [Google Scholar] [CrossRef] [Green Version]
- Suerth, J.D.; Maetzig, T.; Brugman, M.H.; Heinz, N.; Appelt, J.U.; Kaufmann, K.B.; Schmidt, M.; Grez, M.; Modlich, U.; Baum, C.; et al. Alpharetroviral self-inactivating vectors: Long-term transgene expression in murine hematopoietic cells and low genotoxicity. Mol. Ther. 2012, 20, 1022–1032. [Google Scholar] [CrossRef]
- Suerth, J.D.; Maetzig, T.; Galla, M.; Baum, C.; Schambach, A. Self-inactivating alpharetroviral vectors with a split-packaging design. J. Virol. 2010, 84, 6626–6635. [Google Scholar] [CrossRef]
- Kaufmann, K.B.; Brendel, C.; Suerth, J.D.; Mueller-Kuller, U.; Chen-Wichmann, L.; Schwable, J.; Pahujani, S.; Kunkel, H.; Schambach, A.; Baum, C.; et al. Alpharetroviral vector-mediated gene therapy for X-CGD: Functional correction and lack of aberrant splicing. Mol. Ther. 2013, 21, 648–661. [Google Scholar] [CrossRef]
- Cattoglio, C.; Facchini, G.; Sartori, D.; Antonelli, A.; Miccio, A.; Cassani, B.; Schmidt, M.; von Kalle, C.; Howe, S.; Thrasher, A.J.; et al. Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood 2007, 110, 1770–1778. [Google Scholar] [CrossRef]
- Cattoglio, C.; Maruggi, G.; Bartholomae, C.; Malani, N.; Pellin, D.; Cocchiarella, F.; Magnani, Z.; Ciceri, F.; Ambrosi, A.; von Kalle, C.; et al. High-definition mapping of retroviral integration sites defines the fate of allogeneic T cells after donor lymphocyte infusion. PLoS One 2010, 5, e15688. [Google Scholar] [CrossRef]
- Cavazza, A.; Cocchiarella, F.; Bartholomae, C.; Schmidt, M.; Pincelli, C.; Larcher, F.; Mavilio, F. Self-inactivating MLV vectors have a reduced genotoxic profile in human epidermal keratinocytes. Gene Ther. 2013. [Google Scholar] [CrossRef]
- Moiani, A.; Miccio, A.; Rizzi, E.; Severgnini, M.; Pellin, D.; Suerth, J.D.; Baum, C.; de Bellis, G.; Mavilio, F. Deletion of the LTR enhancer/promoter has no impact on the integration profile of MLV vectors in human hematopoietic progenitors. PLoS One 2013, 8, e55721. [Google Scholar]
- Kim, S.Y.; Pritchard, J.K. Adaptive evolution of conserved noncoding elements in mammals. PLoS Genet. 2007, 3, 1572–1586. [Google Scholar]
- John, S.; Sabo, P.J.; Canfield, T.K.; Lee, K.; Vong, S.; Weaver, M.; Wang, H.; Vierstra, J.; Reynolds, A.P.; Thurman, R.E.; et al. Genome-scale mapping of DNase I hypersensitivity. Curr. Protoc. Mol. Biol. 2013. [Google Scholar] [CrossRef]
- Ye, T.; Krebs, A.R.; Choukrallah, M.A.; Keime, C.; Plewniak, F.; Davidson, I.; Tora, L. Seqminer: An integrated chip-seq data interpretation platform. Nucleic Acids Res. 2010, 39, e35. [Google Scholar]
- Ferrigno, O.; Virolle, T.; Djabari, Z.; Ortonne, J.P.; White, R.J.; Aberdam, D. Transposable b2 sine elements can provide mobile rna polymerase ii promoters. Nat. Genet. 2001, 28, 77–81. [Google Scholar]
- Lunyak, V.V.; Prefontaine, G.G.; Nunez, E.; Cramer, T.; Ju, B.G.; Ohgi, K.A.; Hutt, K.; Roy, R.; Garcia-Diaz, A.; Zhu, X.; et al. Developmentally regulated activation of a SINE b2 repeat as a domain boundary in organogenesis. Science 2007, 317, 248–251. [Google Scholar] [CrossRef]
- Biasco, L.; Ambrosi, A.; Pellin, D.; Bartholomae, C.; Brigida, I.; Roncarolo, M.G.; di Serio, C.; von Kalle, C.; Schmidt, M.; Aiuti, A. Integration profile of retroviral vector in gene therapy treated patients is cell-specific according to gene expression and chromatin conformation of target cell. EMBO Mol. Med. 2011, 3, 89–101. [Google Scholar] [CrossRef]
- Berry, C.; Hannenhalli, S.; Leipzig, J.; Bushman, F.D. Selection of target sites for mobile DNA integration in the human genome. PLoS Comput. Biol. 2006, 2, e157. [Google Scholar]
- Lewinski, M.K.; Yamashita, M.; Emerman, M.; Ciuffi, A.; Marshall, H.; Crawford, G.; Collins, F.; Shinn, P.; Leipzig, J.; Hannenhalli, S.; et al. Retroviral DNA integration: Viral and cellular determinants of target-site selection. PLoS Pathog. 2006, 2, e60. [Google Scholar] [CrossRef]
- Cesana, D.; Sgualdino, J.; Rudilosso, L.; Merella, S.; Naldini, L.; Montini, E. Whole transcriptome characterization of aberrant splicing events induced by lentiviral vector integrations. J. Clin. Invest. 2012, 122, 1667–1676. [Google Scholar] [CrossRef]
- Moiani, A.; Paleari, Y.; Sartori, D.; Mezzadra, R.; Miccio, A.; Cattoglio, C.; Cocchiarella, F.; Lidonnici, M.R.; Ferrari, G.; Mavilio, F. Lentiviral vector integration in the human genome induces alternative splicing and generates aberrant transcripts. J. Clin. Invest. 2012, 122, 1653–1666. [Google Scholar] [Green Version]
- Ambrosi, A.; Glad, I.K.; Pellin, D.; Cattoglio, C.; Mavilio, F.; di Serio, C.; Frigessi, A. Estimated comparative integration hotspots identify different behaviors of retroviral gene transfer vectors. PLoS Comput. Biol. 2011, 7, e1002292. [Google Scholar] [CrossRef]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005, 11, 1287–1289. [Google Scholar]
- Llano, M.; Vanegas, M.; Fregoso, O.; Saenz, D.; Chung, S.; Peretz, M.; Poeschla, E.M. LEDGF/p75 determines cellular trafficking of diverse lentiviral but not murine oncoretroviral integrase proteins and is a component of functional lentiviral preintegration complexes. J. Virol. 2004, 78, 9524–9537. [Google Scholar] [CrossRef]
- De Rijck, J.; de Kogel, C.; Demeulemeester, J.; Vets, S.; El Ashkar, S.; Malani, N.; Bushman, F.D.; Landuyt, B.; Husson, S.J.; Busschots, K.; et al. The BET family of proteins targets Moloney murine leukemia virus integration near transcription start sites. Cell Rep. 2013, 5, 886–894. [Google Scholar] [CrossRef]
- Gupta, S.S.; Maetzig, T.; Maertens, G.N.; Sharif, A.; Rothe, M.; Weidner-Glunde, M.; Galla, M.; Schambach, A.; Cherepanov, P.; Schulz, T.F. Bromo- and extraterminal domain chromatin regulators serve as cofactors for murine leukemia virus integration. J. Virol. 2013, 87, 12721–12736. [Google Scholar]
- Sharma, A.; Larue, R.C.; Plumb, M.R.; Malani, N.; Male, F.; Slaughter, A.; Kessl, J.J.; Shkriabai, N.; Coward, E.; Aiyer, S.S.; et al. BET proteins promote efficient murine leukemia virus integration at transcription start sites. Proc. Natl. Acad. Sci. USA 2013, 110, 12036–12041. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Moiani, A.; Suerth, J.D.; Gandolfi, F.; Rizzi, E.; Severgnini, M.; De Bellis, G.; Schambach, A.; Mavilio, F. Genome-Wide Analysis of Alpharetroviral Integration in Human Hematopoietic Stem/Progenitor Cells. Genes 2014, 5, 415-429. https://doi.org/10.3390/genes5020415
Moiani A, Suerth JD, Gandolfi F, Rizzi E, Severgnini M, De Bellis G, Schambach A, Mavilio F. Genome-Wide Analysis of Alpharetroviral Integration in Human Hematopoietic Stem/Progenitor Cells. Genes. 2014; 5(2):415-429. https://doi.org/10.3390/genes5020415
Chicago/Turabian StyleMoiani, Arianna, Julia Debora Suerth, Francesco Gandolfi, Ermanno Rizzi, Marco Severgnini, Gianluca De Bellis, Axel Schambach, and Fulvio Mavilio. 2014. "Genome-Wide Analysis of Alpharetroviral Integration in Human Hematopoietic Stem/Progenitor Cells" Genes 5, no. 2: 415-429. https://doi.org/10.3390/genes5020415