
Nucleosome Dancing at the Tempo of Histone Tail Acetylation

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Nucleosome: An Impediment for Transcription Factor Binding
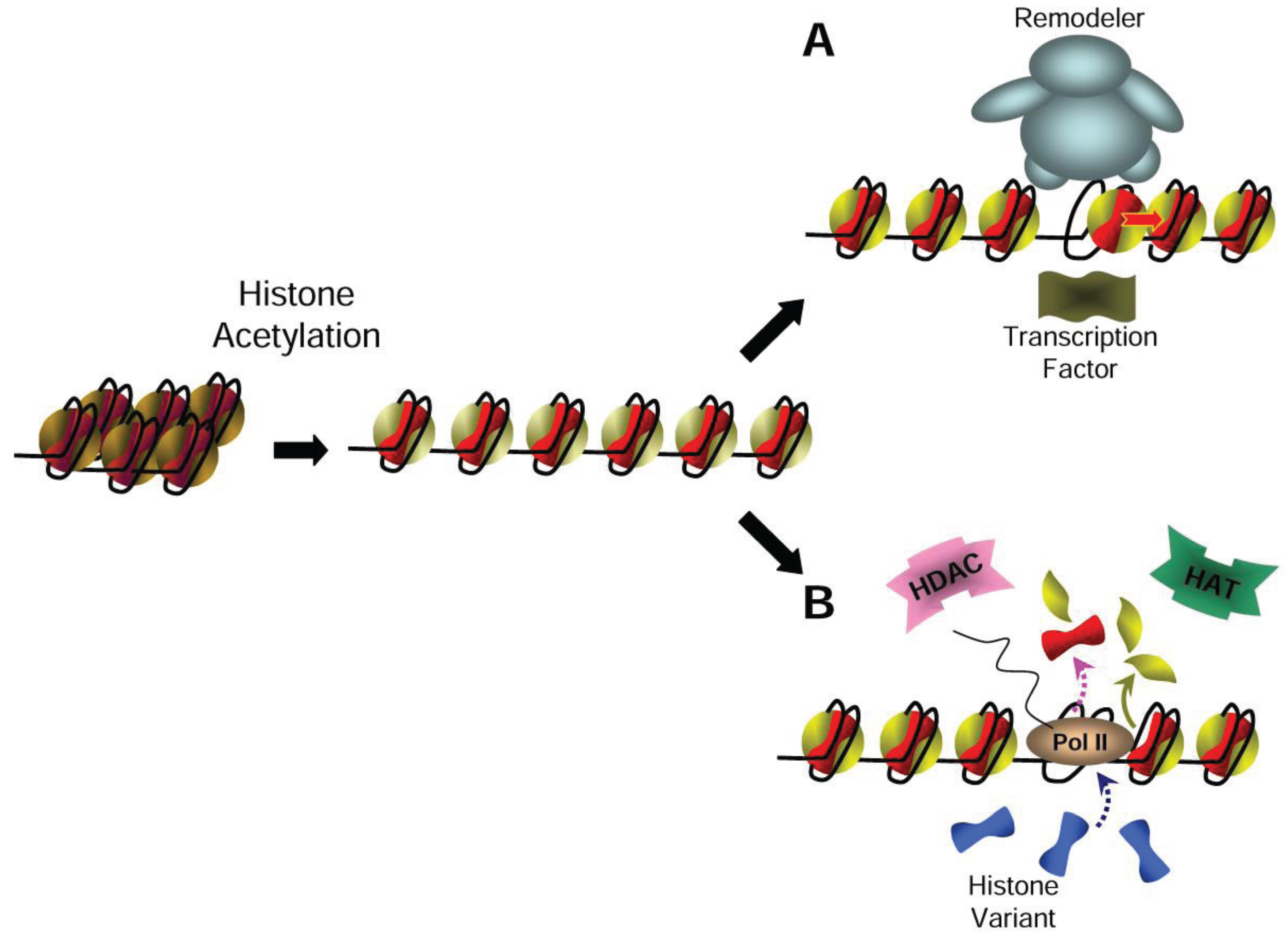
3. Relieving the Nucleosome Barrier by Histone Acetylation
4. Relaxing the Chromatin Structure with Nucleosome Acetylation
5. Painting Chromatin with Histone Tail Acetylation
6. Histone Dynamics in Transcription
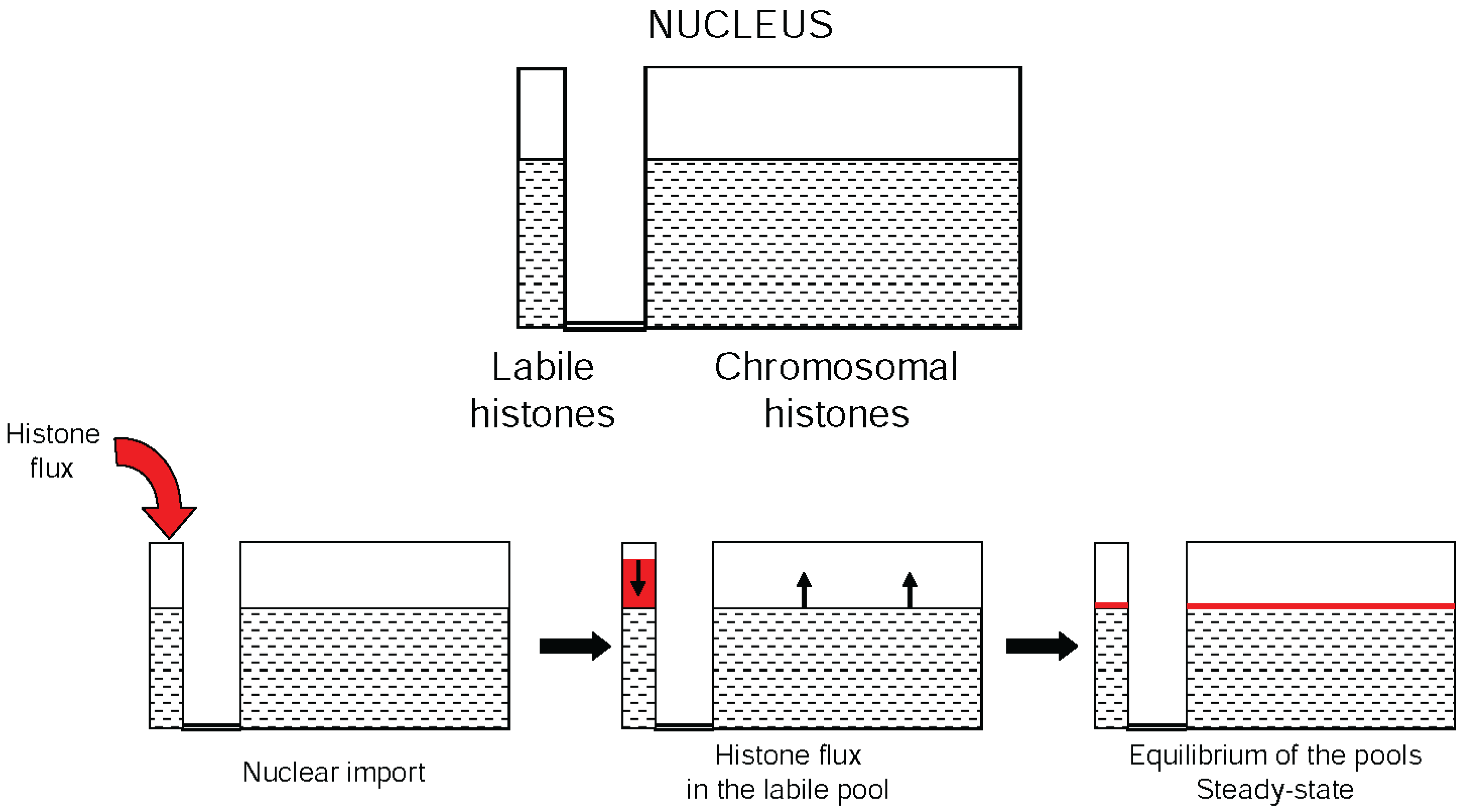
7. The Dynamic Equilibrium of Histone Acetylation
8. Histone Acetylation Coordinates Chromatin Dynamics


9. Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 a resolution. Nature 1997, 389, 251–260. [Google Scholar] [PubMed]
- Lachner, M.; Jenuwein, T. The many faces of histone lysine methylation. Curr. Opin. Cell Biol. 2002, 14, 286–298. [Google Scholar] [CrossRef]
- Lachner, M.; O’Carroll, D.; Rea, S.; Mechtler, K.; Jenuwein, T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001, 410, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.L. ATP-dependent chromatin remodeling: Going mobile. FEBS Lett. 2000, 476, 68–72. [Google Scholar] [CrossRef]
- Cairns, B.R. Chromatin remodeling complexes: Strength in diversity, precision through specialization. Curr. Opin. Genet. Dev. 2005, 15, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Langst, G.; Becker, P.B. Nucleosome mobilization and positioning by ISWI-containing chromatin-remodeling factors. J. Cell Sci. 2001, 114, 2561–2568. [Google Scholar] [PubMed]
- Muchardt, C.; Bourachot, B.; Reyes, J.C.; Yaniv, M. Ras transformation is associated with decreased expression of the brm/SNF2alpha ATPase from the mammalian SWI-SNF complex. EMBO J. 1998, 17, 223–231. [Google Scholar] [CrossRef] [PubMed]
- De la Serna, I.L.; Roy, K.; Carlson, K.A.; Imbalzano, A.N. Myod can induce cell cycle arrest but not muscle differentiation in the presence of dominant negative SWI/SNF chromatin remodeling enzymes. J. Biol. Chem. 2001, 276, 41486–41491. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.F.; Peterson, C.L. A role for the yeast SWI/SNF complex in DNA replication. Nucleic Acids Res. 1999, 27, 2022–2028. [Google Scholar] [CrossRef] [PubMed]
- Thiriet, C.; Hayes, J.J. Chromatin in need of a fix: Phosphorylation of H2AX connects chromatin to DNA repair. Mol. Cell 2005, 18, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Balliano, A.; Hayes, J.J. Mechanism(s) of SWI/SNF-induced nucleosome mobilization. Chembiochem 2011, 12, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Kapros, T.; Bogre, L.; Nemeth, K.; Bako, L.; Gyorgyey, J.; Wu, S.C.; Dudits, D. Differential expression of histone H3 gene variants during cell cycle and somatic embryogenesis in alfalfa. Plant Physiol. 1992, 98, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Loyola, A.; Almouzni, G. Marking histone H3 variants: How, when and why? Trends Biochem. Sci. 2007, 32, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Pilch, D.R.; Sedelnikova, O.A.; Redon, C.; Celeste, A.; Nussenzweig, A.; Bonner, W.M. Characteristics of gamma-H2AX foci at DNA double-strand breaks sites. Biochem. Cell Biol. 2003, 81, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.M.; Henikoff, S. Histone variants: Dynamic punctuation in transcription. Genes Dev. 2014, 28, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Felsenfeld, G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007, 21, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.J.; Wolffe, A.P. The interaction of transcription factors with nucleosomal DNA. Bioessays 1992, 14, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.C.; Workman, J.L. Binding of disparate transcriptional activators to nucleosomal DNA is inherently cooperative. Mol. Cell. Biol. 1995, 15, 1405–1421. [Google Scholar] [PubMed]
- Utley, R.T.; Cote, J.; Owen-Hughes, T.; Workman, J.L. Swi/snf stimulates the formation of disparate activator-nucleosome complexes but is partially redundant with cooperative binding. J. Biol. Chem. 1997, 272, 12642–12649. [Google Scholar] [CrossRef] [PubMed]
- Polach, K.J.; Widom, J. A model for the cooperative binding of eukaryotic regulatory proteins to nucleosomal target sites. J. Mol. Biol. 1996, 258, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Polach, K.J.; Widom, J. Mechanism of protein access to specific DNA sequences in chromatin: A dynamic equilibrium model for gene regulation. J. Mol. Biol. 1995, 254, 130–149. [Google Scholar] [CrossRef] [PubMed]
- Polach, K.J.; Lowary, P.T.; Widom, J. Effects of core histone tail domains on the equilibrium constants for dynamic DNA site accessibility in nucleosomes. J. Mol. Biol. 2000, 298, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Hayes, J.J.; Pruss, D.; Wolffe, A.P. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell 1993, 72, 73–84. [Google Scholar] [CrossRef]
- Vitolo, J.M.; Thiriet, C.; Hayes, J.J. The H3-H4 N-terminal tail domains are the primary mediators of transcription factor IIIA access to 5S DNA within a nucleosome. Mol. Cell. Biol. 2000, 20, 2167–2175. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Schroth, G.P.; Matthews, H.R.; Yau, P.; Bradbury, E.M. Studies of the DNA binding properties of histone H4 amino terminus. Thermal denaturation studies reveal that acetylation markedly reduces the binding constant of the H4 “tail” to DNA. J. Biol. Chem. 1993, 268, 305–314. [Google Scholar] [PubMed]
- Wang, X.; Moore, S.C.; Laszckzak, M.; Ausio, J. Acetylation increases the alpha-helical content of the histone tails of the nucleosome. J. Biol. Chem. 2000, 275, 35013–35020. [Google Scholar] [CrossRef] [PubMed]
- Vidali, G.; Boffa, L.C.; Bradbury, E.M.; Allfrey, V.G. Butyrate suppression of histone deacetylation leads to accumulation of multiacetylated forms of histones H3 and H4 and increased DNase I sensitivity of the associated DNA sequences. Proc. Natl. Acad. Sci. USA 1978, 75, 2239–2243. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hayes, J.J. Acetylation mimics within individual core histone tail domains indicate distinct roles in regulating the stability of higher-order chromatin structure. Mol. Cell. Biol. 2008, 28, 227–236. [Google Scholar] [CrossRef]
- Wu, C.; Wong, Y.C.; Elgin, S.C. The chromatin structure of specific genes: II. Disruption of chromatin structure during gene activity. Cell 1979, 16, 807–814. [Google Scholar] [CrossRef]
- Wu, C. The 5' ends of drosophila heat shock genes in chromatin are hypersensitive to DNase I. Nature 1980, 286, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Korber, P.; Barbaric, S. The yeast PHO5 promoter: From single locus to systems biology of a paradigm for gene regulation through chromatin. Nucleic Acids Res. 2014, 42, 10888–10902. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.D.; Wagner, K.; Horz, W. Histone acetylation and chromatin remodeling. Exp. Cell Res. 2001, 265, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Reinke, H.; Gregory, P.D.; Horz, W. A transient histone hyperacetylation signal marks nucleosomes for remodeling at the PHO8 promoter in vivo. Mol. Cell 2001, 7, 529–538. [Google Scholar] [CrossRef]
- Syntichaki, P.; Topalidou, I.; Thireos, G. The gcn5 bromodomain co-ordinates nucleosome remodelling. Nature 2000, 404, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [PubMed]
- Zeng, L.; Zhou, M.M. Bromodomain: An acetyl-lysine binding domain. FEBS Lett. 2002, 513, 124–128. [Google Scholar] [CrossRef]
- Jeanmougin, F.; Wurtz, J.M.; Le Douarin, B.; Chambon, P.; Losson, R. The bromodomain revisited. Trends Biochem. Sci. 1997, 22, 151–153. [Google Scholar] [CrossRef]
- Ornaghi, P.; Ballario, P.; Lena, A.M.; Gonzalez, A.; Filetici, P. The bromodomain of Gcn5p interacts in vitro with specific residues in the N terminus of histone H4. J. Mol. Biol. 1999, 287, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.M.; Birley, A.J.; Lavender, J. Histone H4 isoforms acetylated at specific lysine residues define individual chromosomes and chromatin domains in drosophila polytene nuclei. Cell 1992, 69, 375–384. [Google Scholar] [CrossRef]
- Kireeva, M.L.; Walter, W.; Tchernajenko, V.; Bondarenko, V.; Kashlev, M.; Studitsky, V.M. Nucleosome remodeling induced by RNA polymerase II: Loss of the H2A/H2B dimer during transcription. Mol. Cell 2002, 9, 541–552. [Google Scholar] [CrossRef]
- Belotserkovskaya, R.; Oh, S.; Bondarenko, V.A.; Orphanides, G.; Studitsky, V.M.; Reinberg, D. Fact facilitates transcription-dependent nucleosome alteration. Science 2003, 301, 1090–1093. [Google Scholar] [CrossRef] [PubMed]
- Thiriet, C.; Hayes, J.J. Replication-independent core histone dynamics at transcriptionally active loci in vivo. Genes Dev. 2005, 19, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.; Henikoff, S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell 2002, 9, 1191–1200. [Google Scholar] [CrossRef]
- Schwartz, B.E.; Ahmad, K. Transcriptional activation triggers deposition and removal of the histone variant h3.3. Genes Dev. 2005, 19, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Mito, Y.; Henikoff, J.G.; Henikoff, S. Genome-scale profiling of histone H3.3 replacement patterns. Nat. Genet. 2005, 37, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Thiriet, C.; Hayes, J.J. Histone dynamics during transcription: Exchange of H2A/H2B dimers and H3/H4 tetramers during pol II elongation. Results Probl. Cell Differ. 2006, 41, 77–90. [Google Scholar] [PubMed]
- McKittrick, E.; Gafken, P.R.; Ahmad, K.; Henikoff, S. Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc. Natl. Acad. Sci. USA 2004, 101, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Gorovsky, M.A. Constitutive expression, not a particular primary sequence, is the important feature of the H3 replacement variant HV2 in Tetrahymena thermophila. Mol. Cell. Biol. 1997, 17, 6303–6310. [Google Scholar] [PubMed]
- Cui, B.; Liu, Y.; Gorovsky, M.A. Deposition and function of histone H3 variants in Tetrahymena thermophila. Mol. Cell. Biol. 2006, 26, 7719–7730. [Google Scholar] [CrossRef] [PubMed]
- Hodl, M.; Basler, K. Transcription in the absence of histone H3.3. Curr. Biol. 2009, 19, 1221–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szenker, E.; Ray-Gallet, D.; Almouzni, G. The double face of the histone variant H3.3. Cell Res. 2011, 21, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Jamai, A.; Imoberdorf, R.M.; Strubin, M. Continuous histone H2B and transcription-dependent histone H3 exchange in yeast cells outside of replication. Mol. Cell 2007, 25, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Litt, M.D.; Simpson, M.; Recillas-Targa, F.; Prioleau, M.N.; Felsenfeld, G. Transitions in histone acetylation reveal boundaries of three separately regulated neighboring loci. EMBO J. 2001, 20, 2224–2235. [Google Scholar] [CrossRef] [PubMed]
- Fromm, G.; de Vries, C.; Byron, R.; Fields, J.; Fiering, S.; Groudine, M.; Bender, M.A.; Palis, J.; Bulger, M. Histone hyperacetylation within the beta-globin locus is context-dependent and precedes high-level gene expression. Blood 2009, 114, 3479–3488. [Google Scholar] [CrossRef] [PubMed]
- Bulger, M. Hyperacetylated chromatin domains: Lessons from heterochromatin. J. Biol. Chem. 2005, 280, 21689–21692. [Google Scholar] [CrossRef] [PubMed]
- Bulger, M.; Sawado, T.; Schubeler, D.; Groudine, M. Chips of the beta-globin locus: Unraveling gene regulation within an active domain. Curr. Opin. Genet. Dev. 2002, 12, 170–177. [Google Scholar] [CrossRef]
- Wang, X.; Hayes, J.J. Site-specific binding affinities within the H2B tail domain indicate specific effects of lysine acetylation. J. Biol. Chem. 2007, 282, 32867–32876. [Google Scholar] [CrossRef] [PubMed]
- Chahal, S.S.; Matthews, H.R.; Bradbury, E.M. Acetylation of histone H4 and its role in chromatin structure and function. Nature 1980, 287, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Waterborg, J.H.; Matthews, H.R. Intranuclear localization of histone acetylation in Physarum polycephalum and the structure of functionally active chromatin. Cell Biophys. 1983, 5, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Waterborg, J.H.; Matthews, H.R. Patterns of histone acetylation in the cell cycle of Physarum polycephalum. Biochemistry 1983, 22, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Waterborg, J.H.; Matthews, H.R. Patterns of histone acetylation in Physarum polycephalum. H2a and h2b acetylation is functionally distinct from h3 and h4 acetylation. Eur. J. Biochem. 1984, 142, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Waterborg, J.H. Dynamics of histone acetylation in Chlamydomonas reinhardtii. J. Biol. Chem. 1998, 273, 27602–27609. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rodas, G.; Brosch, G.; Golderer, G.; Lindner, H.; Grobner, P.; Loidl, P. Enzymes involved in the dynamic equilibrium of core histone acetylation of Physarum polycephalum. FEBS Lett. 1992, 296, 82–86. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Elliott, O.G.; Murphy, K.; Hayes, J.J.; Thiriet, C. Replication-independent nucleosome exchange is enhanced by local and specific acetylation of histone H4. Nucleic Acids Res. 2013, 41, 2228. [Google Scholar] [CrossRef] [PubMed]
- Gurard-Levin, Z.A.; Quivy, J.P.; Almouzni, G. Histone chaperones: Assisting histone traffic and nucleosome dynamics. Annu. Rev. Biochem. 2014, 83, 487–517. [Google Scholar] [CrossRef] [PubMed]
- Gunjan, A.; Paik, J.; Verreault, A. Regulation of histone synthesis and nucleosome assembly. Biochimie 2005, 87, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Kulaeva, O.I.; Gaykalova, D.A.; Studitsky, V.M. Transcription through chromatin by RNA polymerase II: Histone displacement and exchange. Mutat. Res. 2007, 618, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Cook, P.R. Kinetics of core histones in living human cells: Little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 2001, 153, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Tropberger, P.; Pott, S.; Keller, C.; Kamieniarz-Gdula, K.; Caron, M.; Richter, F.; Li, G.; Mittler, G.; Liu, E.T.; Buhler, M.; et al. Regulation of transcription through acetylation of H3K122 on the lateral surface of the histone octamer. Cell 2013, 152, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.; Hancock, S.M.; Buning, R.; Routh, A.; Chapman, L.; Somers, J.; Owen-Hughes, T.; van Noort, J.; Rhodes, D.; Chin, J.W. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol. Cell 2009, 36, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Smolle, M.; Li, H.; Gogol, M.M.; Saint, M.; Kumar, S.; Natarajan, K.; Workman, J.L. Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature 2012, 489, 452–455. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galvani, A.; Thiriet, C. Nucleosome Dancing at the Tempo of Histone Tail Acetylation. Genes 2015, 6, 607-621. https://doi.org/10.3390/genes6030607
Galvani A, Thiriet C. Nucleosome Dancing at the Tempo of Histone Tail Acetylation. Genes. 2015; 6(3):607-621. https://doi.org/10.3390/genes6030607
Chicago/Turabian StyleGalvani, Angélique, and Christophe Thiriet. 2015. "Nucleosome Dancing at the Tempo of Histone Tail Acetylation" Genes 6, no. 3: 607-621. https://doi.org/10.3390/genes6030607